OTU clustering
Overview
Teaching: 15 min
Exercises: 30 minQuestions
Is it better to denoise or cluster sequence data?
Objectives
Learn to cluster sequences into OTUs with the VSEARCH pipeline
OTU clustering
Introduction
Now that the dataset is filtered and each sequence is assigned to their corresponding sample, we are ready for the next step in the bioinformatic pipeline, i.e., looking for biologically meaningful or biologically correct sequences. This is done through denoising or clustering the dataset, while at the same time removing any chimeric sequences and remaining PCR and/or sequencing errors.
There is still an ongoing debate on what the best approach is to obtain these biologically meaningful sequences. If you would like more information, these are two good papers to start with paper 1 (EMP); paper 2 (EMP). We won’t be discussing this topic in much detail during the workshop, but this is the basic idea…
When clustering the dataset, OTUs (Operational Taxonomic Units) will be generated by combining sequences that are similar to a set percentage level (traditionally 97%), with the most abundant sequence identified as the true sequence. When clustering at 97%, this means that if a sequence is more than 3% different than the generated OTU, a second OTU will be generated. The concept of OTU clustering was introduced in the 1960s and has been debated since. Clustering the dataset is usually used for identifying species in metabarcoding data.
Denoising, on the other hand, attempts to identify all correct biological sequences through an algorithm, which his visualised in the figure below. In short, denoising will cluster the data with a 100% threshold and tries to identify errors based on abundance differences. The retained sequences are called ZOTU (Zero-radius Operation Taxonomic Unit) or ASVs (Amplicon Sequence Variants). Denoising the dataset is usually used for identifying intraspecific variation in metabarcoding data.
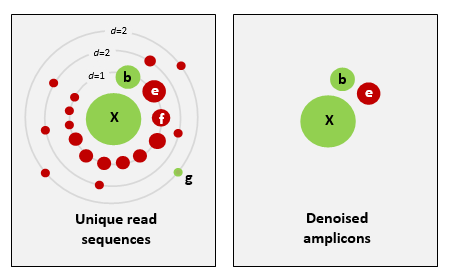
This difference in approach may seem small but has a very big impact on your final dataset!
When you denoise the dataset, it is expected that one species may have more than one ZOTU, while if you cluster the dataset, it is expected that an OTU may have more than one species assigned to it. This means that you may lose some correct biological sequences that are present in your data when you cluster the dataset, because they will be clustered together. In other words, you will miss out on differentiating closely related species and intraspecific variation. For denoising, on the other hand, it means that when the reference database is incomplete, or you plan to work with ZOTUs instead of taxonomic assignments, your diversity estimates will be highly inflated.
For this workshop, we will follow the clustering pathway, since most people are working on vertebrate datasets with good reference databases and are interested in taxonomic assignments.
Dereplicating the data
Before we can cluster our dataset, we need to dereplicate the sequences into unique sequences. Since metabarcoding data is based on an amplification method, the same DNA molecule can be sequenced several times. In order to reduce file size and computational time, it is convenient to work with unique sequences.
During dereplication, we will remove unique sequences that are not prevalent in our dataset. There is still not a standardised guide to the best cutoff. Many eDNA metabarcoding publications remove only the singleton sequences, i.e., sequences that are present once in the dataset, while others opt to remove unique sequences occurring less than three or ten times. Given there isn’t a standard approach, we will be using the more strict approach of removing sequences occurring fewer than 10 times in the dataset. Lastly, we will sort the sequences based on abundance, as this will be needed for the OTU clustering step.
nano derep
#!/bin/bash
source moduleload
cd ../output/
vsearch --derep_fulllength filtered.fasta --relabel uniq. --output uniques.fasta --sizeout --minuniquesize 10
vsearch --sortbysize uniques.fasta --output sorted_uniques.fasta
chmod +x derep
./derep
vsearch v2.17.1_linux_x86_64, 503.8GB RAM, 72 cores
https://github.com/torognes/vsearch
Dereplicating file filtered.fasta 100%
25083932 nt in 123537 seqs, min 176, max 214, avg 203
Sorting 100%
6645 unique sequences, avg cluster 18.6, median 1, max 19554
Writing output file 100%
286 uniques written, 6359 clusters discarded (95.7%)
vsearch v2.17.1_linux_x86_64, 503.8GB RAM, 72 cores
https://github.com/torognes/vsearch
Reading file uniques.fasta 100%
58019 nt in 286 seqs, min 189, max 213, avg 203
Getting sizes 100%
Sorting 100%
Median abundance: 17
Writing output 100%
To check if everything executed properly, we can list the files that are in the output folder and check if both output files have been generated (uniques.fasta and sorted_uniques.fasta).
$ ls -ltr ../output/
-rw-rw----+ 1 jeuge18p uoo02328 54738505 Nov 17 13:51 relabel.fastq
-rw-rw----+ 1 jeuge18p uoo02328 27440390 Nov 17 14:31 filtered.fasta
-rw-rw----+ 1 jeuge18p uoo02328 52647859 Nov 17 14:31 filtered.fastq
-rw-rw----+ 1 jeuge18p uoo02328 63983 Nov 17 15:13 uniques.fasta
-rw-rw----+ 1 jeuge18p uoo02328 63983 Nov 17 15:13 sorted_uniques.fasta
We can also check how these commands have altered our files. Let’s have a look at the sorted_uniques.fasta file, by using the head command.
$ head ../output/sorted_uniques.fasta
>uniq.1;size=19554
TTTAGAACAGACCATGTCAGCTACCCCCTTAAACAAGTAGTAATTATTGAACCCCTGTTCCCCTGTCTTTGGTTGGGGCG
ACCACGGGGAAGAAAAAAACCCCCACGTGGACTGGGAGCACCTTACTCCTACAACTACGAGCCACAGCTCTAATGCGCAG
AATTTCTGACCATAAGATCCGGCAAAGCCGATCAACGGACCG
>uniq.2;size=14321
ACTAAGGCATATTGTGTCAAATAACCCTAAAACAAAGGACTGAACTGAACAAACCATGCCCCTCTGTCTTAGGTTGGGGC
GACCCCGAGGAAACAAAAAACCCACGAGTGGAATGGGAGCACTGACCTCCTACAACCAAGAGCTGCAGCTCTAACTAATA
GAATTTCTAACCAATAATGATCCGGCAAAGCCGATTAACGAACCA
>uniq.3;size=8743
ACCAAAACAGCTCCCGTTAAAAAGGCCTAGATAAAGACCTATAACTTTCAATTCCCCTGTTTCAATGTCTTTGGTTGGGG
Study Questions 1
What do you think the following parameters are doing?
--sizeout--minuniquesize 2--relabel Uniq.Solution
--sizeout: this will print the number of times a sequence has occurred in the header information. You can use theheadcommand on the sorted file to check how many times the most abundant sequence was encountered in our sequencing data.
--minuniquesize 2: only keeps the dereplicated sequences if they occur twice or more. To check if this actually happened, you can use thetailcommand on the sorted file to see if there are any dereplicates sequences with--sizeout 1.
--relabel Uniq.: this parameter will relabel the header information toUniq.followed by an ascending number, depending on how many different sequences have been encountered.
Chimera removal and clustering
Now that we have dereplicated and sorted our sequences, we can remove chimeric sequences, cluster our data to generate OTUs, and generate a frequency table. We will do this by writing the following script:
nano cluster
#!/bin/bash
source moduleload
cd ../output/
vsearch --uchime3_denovo sorted_uniques.fasta --nonchimeras nonchimeras.fasta
vsearch --cluster_size nonchimeras.fasta --centroids ../final/otus.fasta --relabel otu. --id 0.97
vsearch --usearch_global filtered.fasta --db ../final/otus.fasta --strand plus --otutabout ../final/otutable.txt --id 0.97
chmod +x cluster
./cluster
vsearch v2.17.1_linux_x86_64, 503.8GB RAM, 72 cores
https://github.com/torognes/vsearch
Reading file sorted_uniques.fasta 100%
58019 nt in 286 seqs, min 189, max 213, avg 203
Masking 100%
Sorting by abundance 100%
Counting k-mers 100%
Detecting chimeras 100%
Found 28 (9.8%) chimeras, 258 (90.2%) non-chimeras,
and 0 (0.0%) borderline sequences in 286 unique sequences.
Taking abundance information into account, this corresponds to
885 (0.8%) chimeras, 111140 (99.2%) non-chimeras,
and 0 (0.0%) borderline sequences in 112025 total sequences.
vsearch v2.17.1_linux_x86_64, 503.8GB RAM, 72 cores
https://github.com/torognes/vsearch
Reading file nonchimeras.fasta 100%
52356 nt in 258 seqs, min 189, max 213, avg 203
Masking 100%
Sorting by abundance 100%
Counting k-mers 100%
Clustering 100%
Sorting clusters 100%
Writing clusters 100%
Clusters: 26 Size min 1, max 58, avg 9.9
Singletons: 5, 1.9% of seqs, 19.2% of clusters
vsearch v2.17.1_linux_x86_64, 503.8GB RAM, 72 cores
https://github.com/torognes/vsearch
Reading file ../final/otus.fasta 100%
5277 nt in 26 seqs, min 189, max 213, avg 203
Masking 100%
Counting k-mers 100%
Creating k-mer index 100%
Searching 100%
Matching unique query sequences: 122096 of 123537 (98.83%)
Writing OTU table (classic) 100%
Once successfully executed, we have generated a list of 26 OTUs and an OTU table containing a combined total of 122,096 sequences.
Study Questions 2
The results of the last script creates three files:
nonchimeras.fasta,otus.fasta, andotutable.txt. Let’s have a look at these files to see what the output looks like. From looking at the output, can you guess what some of these VSEARCH parameters are doing?
--sizein--sizeout--centroids--relabelHow about
--fasta_width 0? Try rerunning the script without this command to see if you can tell what this option is doing.Hint: you can always check the help documentation of the module by loading the module
source moduleloadand calling the help documentationvsearch -hor by looking at the manual of the software, which can usually be found on the website.
Optional extra 1
We discussed the difference between denoising and clustering. You can change the last script to have VSEARCH do denoising instead of clustering. Make a new script to run denoising. It is similar to
cluster, except you do not use thevsearch --cluster_sizecommand. Instead you will use:vsearch --cluster_unoise sorted_uniques.fasta --sizein --sizeout --fasta_width 0 --centroids centroids_unoise.fastaYou will still need the commands to remove chimeras and create a frequency table. Make sure to change the names of the files so that you are making a new frequency table and OTU fasta file (maybe call it
asvs.fasta). If you leave the old name in, it will be overwritten when you run this script.Give it a try and compare the number of OTUs between clustering and denoising outputs.
Optional extra 2
The
--idargument is critical for clustering. What happens if you raise or lower this number? Try rerunning the script to see how it changes the results. Note: make sure to rename the output each time or you will not be able to compare to previous runs.
Key Points
Clustering sequences into OTUs can miss closely related species
Denoising sequences can create overestimate diversity when reference databases are incomplete