Quality filtering
Overview
Teaching: 15 min
Exercises: 30 minQuestions
What parameters to use to filter metabarcoding data on quality?
Objectives
Learn to filter sequences on quality using the VSEARCH pipeline
Quality filtering
Introduction
During the quality filtering step, we will discard all sequences that do not adhere to a specific set of rules. We will be using the program VSEARCH (EMP) for this step. During quality filtering, we will filter out all sequences that do not adhere to a minimum and maximum length, have unassigned base calls (‘N’), and have a higher expected error than 1. Once we have filtered our data, we can check the quality with FastQC and compare it to the raw sequencing file.
Filtering sequence data
$ nano qual
$ #!/bin/bash
$
$ source moduleload
$
$ cd ../output/
$
$ vsearch --fastq_filter relabel.fastq --fastq_maxee 1.0 --fastq_maxlen 230 --fastq_minlen 150 --fastq_maxns 0 --fastaout filtered.fasta --fastqout filtered.fastq
$ chmod +x qual
$ ./qual
vsearch v2.17.1_linux_x86_64, 503.8GB RAM, 72 cores
https://github.com/torognes/vsearch
Reading input file 100%
123537 sequences kept (of which 0 truncated), 5337 sequences discarded.
After quality filtering, we have 123,537 sequences left over in the fastq and fasta files. Before we continue with the pipeline, let’s check if the quality filtering was successful using FastQC.
nano qualcheck
#!/bin/bash
source moduleload
cd ../output/
fastqc filtered.fastq -o ../meta/
$ chmod +x qualcheck
$ ./qualcheck
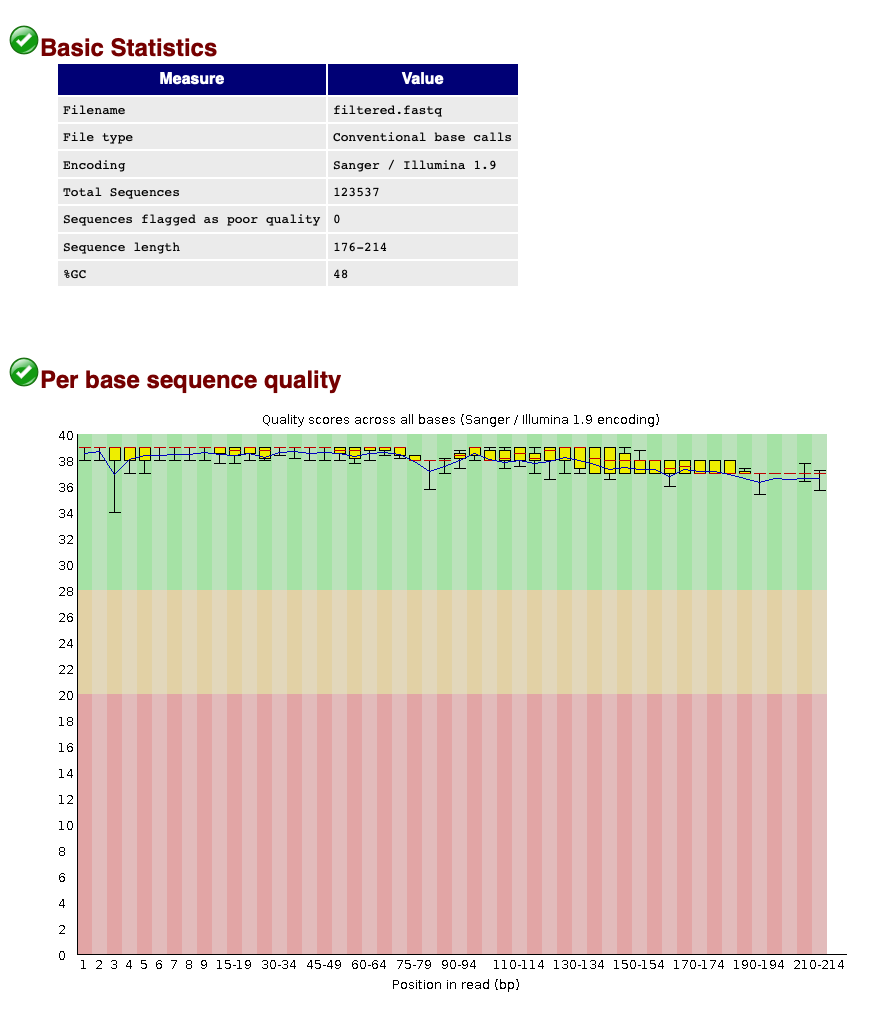
When we now look at the Basic Statistics and Per base sequence quality tabs, we can see that:
- Our total number of sequences has been reduced to 123,537, similar to the number specified by our
qualscript. - The sequence length ranges from 176-214, according to amplicon length and within the range of the parameters chosen in the
qualscript. - The sequence quality is high throughout the amplicon length (within the green zone).
We can, therefore, conclude that quality filtering was successful and we can now move to the clustering step of the pipeline.
Count the number of sequences in a fastq file
before we continue with our bioinformatic pipeline, let’s first determine how to count the number of sequences in a fastq file in the terminal
Hint 1: think about the file structure of the fastq file. Hint 2: think about how to calculate number of sequences in a fasta file
Solution
grep -c "^@" filtered.fastq
Key Points
Quality filtering on sequence length, ambiguous basecalls, and maximum error rate