Nanopore-based sequencing: introduction
Overview
Teaching: 20 min
Exercises: 0 minQuestions
What is nanopore-based sequencing?
Objectives
Understand details of ONT sequencing platforms.
What is this module about?
- Long-read sequencing with the Oxford Nanopore Technologies (ONT) platform.
- WHY?
- recent technology advances have led to a surge in popularly for this platform.
- Huge scope for really exciting (and low cost) science
- Concepts and skills learned here are transferable to other sequencing technologies.
- WHAT?
- Genome assembly
- RNA-seq
- Variant identification (single base & structural)
- Base modification (e.g., methylation)
- Metagenomics
ONT platforms

The MinION
http://www.nature.com/news/data-from-pocket-sized-genome-sequencer-unveiled-1.14724
- It’s a USB stick sequencer!!
- Rough specs (2014)
- 6-8 hour run time
- sequence per run: ~110Mbp
- average read length: 5,400bp
- reads up to 10kbp
- 2022 specs:
- can run for up to 72 hours
- Max yield per run: 50Gbp
- Max read length recorded: >4Mbp

https://nanoporetech.com https://nanoporetech.com/products/specifications
The MinION Mk1B
- The MinION Mk1B is the current version of ONT’s original sequencer.
- Connects to a computer via USB.

https://nanoporetech.com/products/minion

The MinION Mk1C
- The MinION Mk1C provides a truly portable sequencing option, with built in compute and touchscreen.
- For the price, however, the hardware is not particularly impressive.

https://nanoporetech.com/products/minion

The GridION
- The GridION offers a “medium-throughput” option for Nanopore-based sequencing: can run up to five flowcells at once.
- The MinION (Mk1B and Mk1C) and GridION use the same flow cells
- The Otago Genomics Facility has one of these.

https://nanoporetech.com/products/gridion
The Flongle
- The Flongle uses an adapter to allow a smaller (and cheaper) flow cell to be used in the MinION and GridION devices.
- Single-use system provides low-cost option for targeted sequencing (e.g., diagnostic applications).

https://nanoporetech.com/products/flongle
PromethION
- The higher throughout PromethION uses a smaller cartridge-like flow cell. Two options: 24 or 48 flow cells.

https://nanoporetech.com/products/promethion

Coming soon now: the P2

Smaller device (standalone or connect to host computer) that can run Promethion flow cells.
https://nanoporetech.com/products/p2
MinION flowcell characteristics
- MinION flowcells typically have ~1200-1800 active pores (ONT guarantees at least 800 active pores).
- Sequencing occurs at roughly 450 bases per second (ONT recommends keeping speed above 300 bases per second - additional reagents can be added to “refuel” the flowcell*)
-
BUT: pores are not constantly active, and can become blocked during the run
- https://community.nanoporetech.com/protocols/experiment-companion-minknow/v/mke_1013_v1_revbm_11apr2016/refuelling-your-flow-cell
Nanopore technology

https://nanoporetech.com/how-it-works
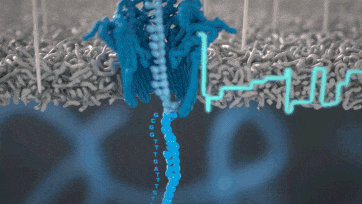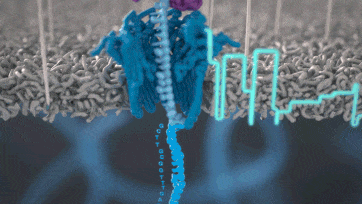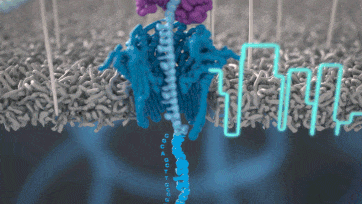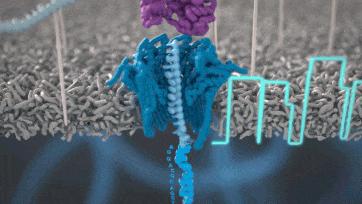
Nanopore technology
- A motor protein (green) passes a strand of DNA through a nanopore (blue). The current is changed as the bases G, A, T and C pass through the pore in different combinations.

https://nanoporetech.com/how-it-works
Nanopore movies
For more detailed information about ONT sequencing:
Technological advances…
- Since its introduction, nanopore sequencing has seen a number of improvements.
- The initial product was realtively slow, expensive (per base sequenced) and error prone (i.e., incorrect bases calls).
- Incremental improvements have led to major advances in both speed and accuracy.
2D sequencing (prior to 2017)
- Hairpin-based approach provided natural error detection methodology:
- Link DNA strands with a hairpin adapter.
- Sequence template strand followed by complement.
- Basecall and compare sequences to produce consensus.
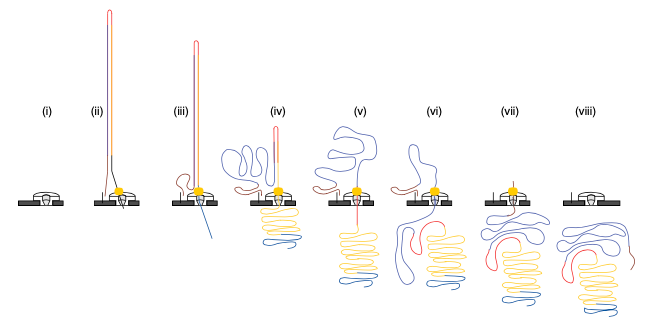
Jain, et al. The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biol 17, 239 (2016). https://doi.org/10.1186/s13059-016-1103-0
What happened to 2D reads?
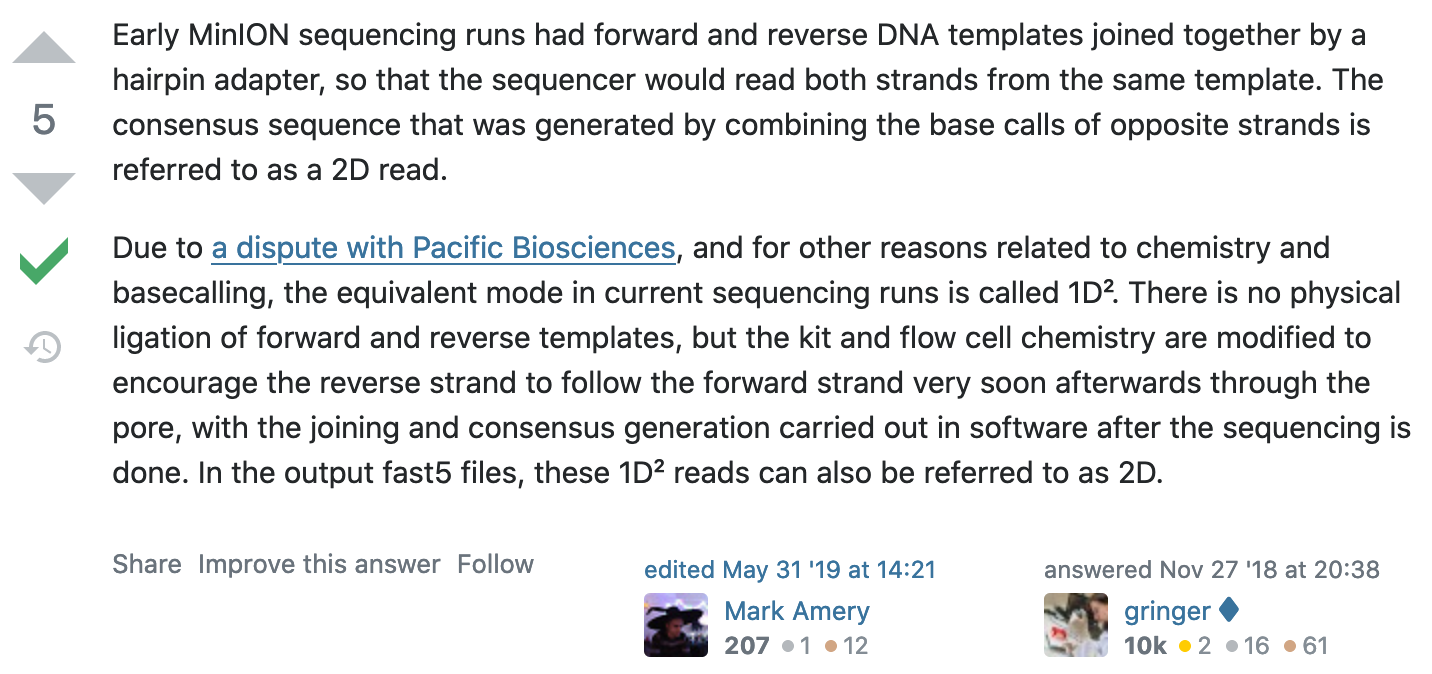
https://bioinformatics.stackexchange.com/questions/5525/what-are-2d-reads-in-the-oxford-minion/5528
The hairpin lawsuit…
- PacBio (competitor in the long-read space) and ONT have filed a number of lawsuits against each other over the past few years.

https://www.genomeweb.com/sequencing/pacbio-oxford-nanopore-settle-patent-dispute-europe
1D$^2$ sequencing
- In 2017 ONT announced the new 1D$^2$ chemistry
- Showed higher accuracy that 1D (and SAID it was better than 2D)
- It didn’t last long…
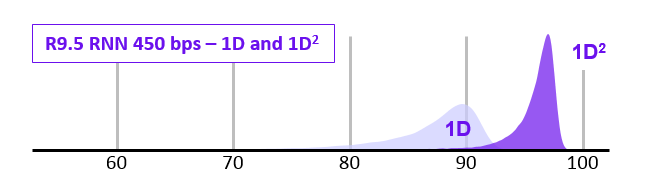
- Video at link below:
https://nanoporetech.com/about-us/news/1d-squared-kit-available-store-boost-accuracy-simple-prep
Pore imporvements: the R10 pore
- ONT introduced the new R10 pore in 2019 (previous was R9.4.1).
- Main differences were longer barrel and dual reader head: gave improved resolution of homopolymer runs.
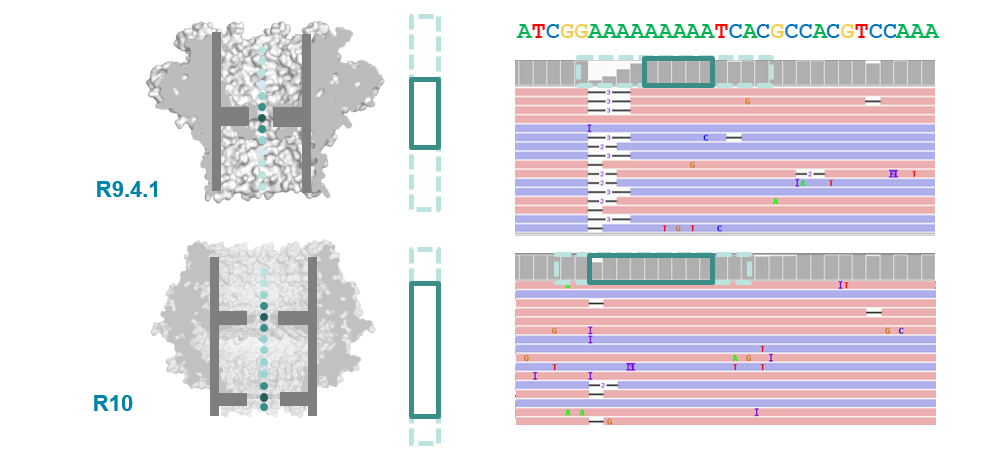
R10.3 vs R9.4.1 performance
- With a bit more tweaking (to get to R10.3) ONT improved 1D (i.e., single-strand) sequencing accuracy, although throughput is still not as high as the R.9.4.1 pore.
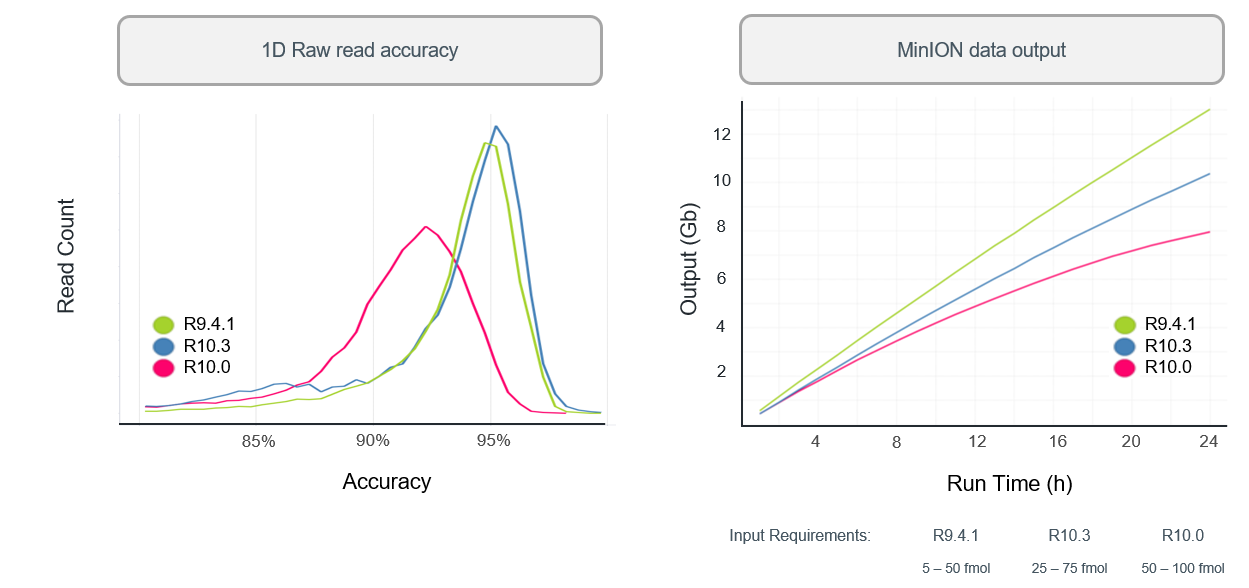
Q20+: the return of 2D reads…
- The previous ONT products are “Q10” (we’ll discuss this soon), meaning that the error rate is roughly 1 incorrect base call per 10 bases (that’s high!)
- The new “Q20+” products are now available - moves to less than 1 error per 100 bases (a little more respectable, but still well below short-read technologies like Illumina).
- Upgrade includes a return to the “2D” approach.

Making improvements
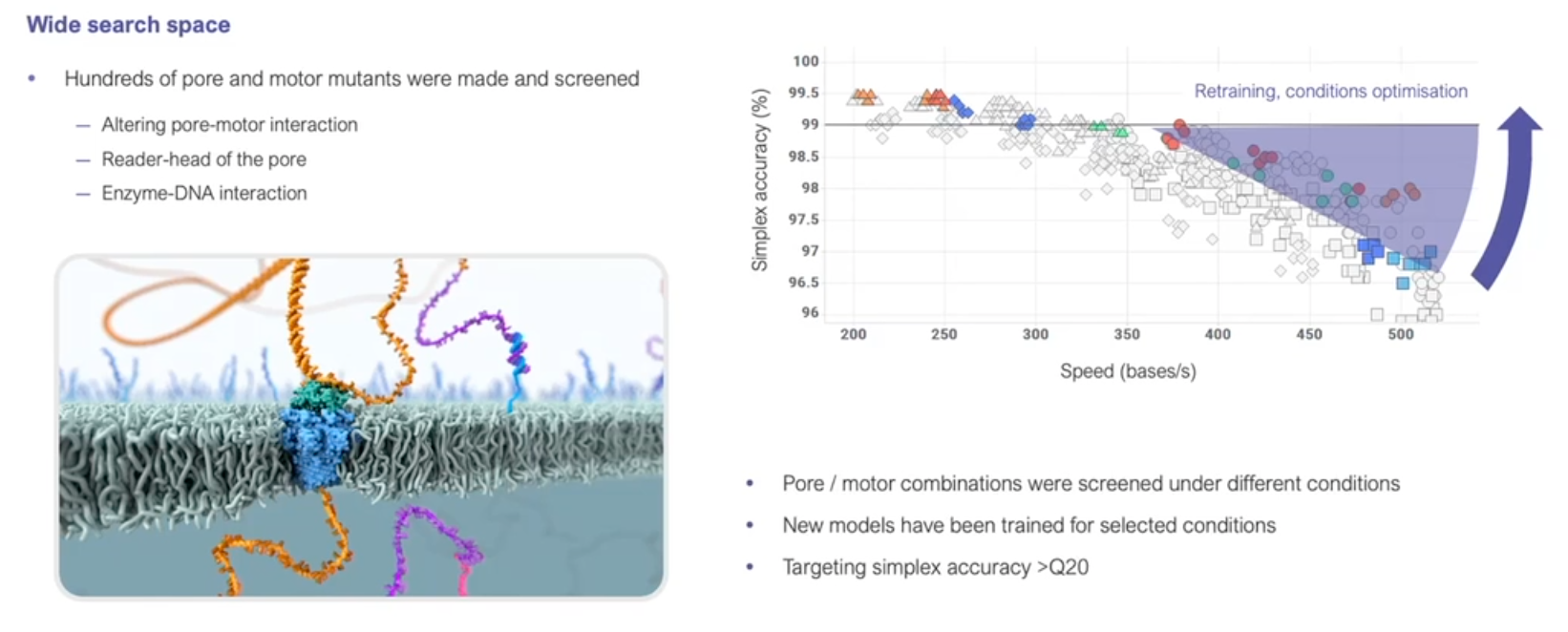
https://nanoporetech.com/resource-centre/london-calling-2022-update-oxford-nanopore-technologies
Q20+ and Kit 14
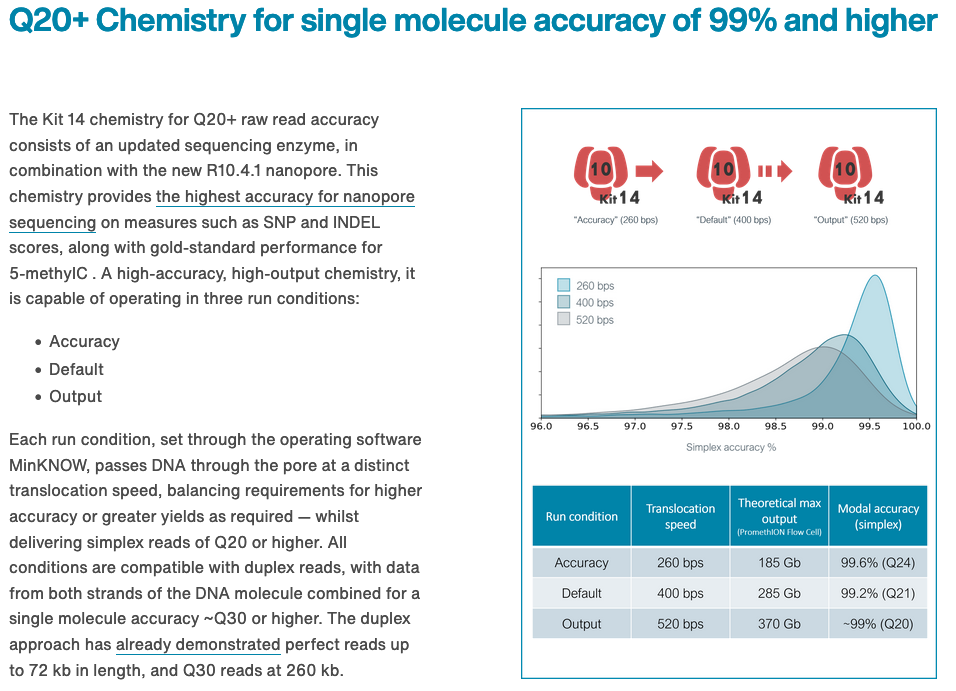
https://nanoporetech.com/q20plus-chemistry
Nanopore accuracy
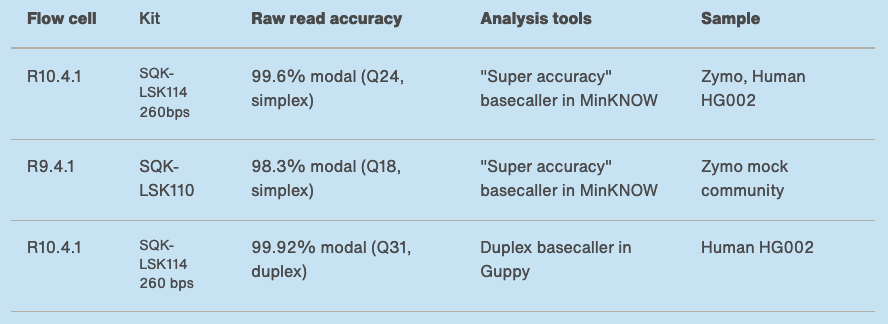
https://nanoporetech.com/accuracy
Duplex (and simplex) accuracy
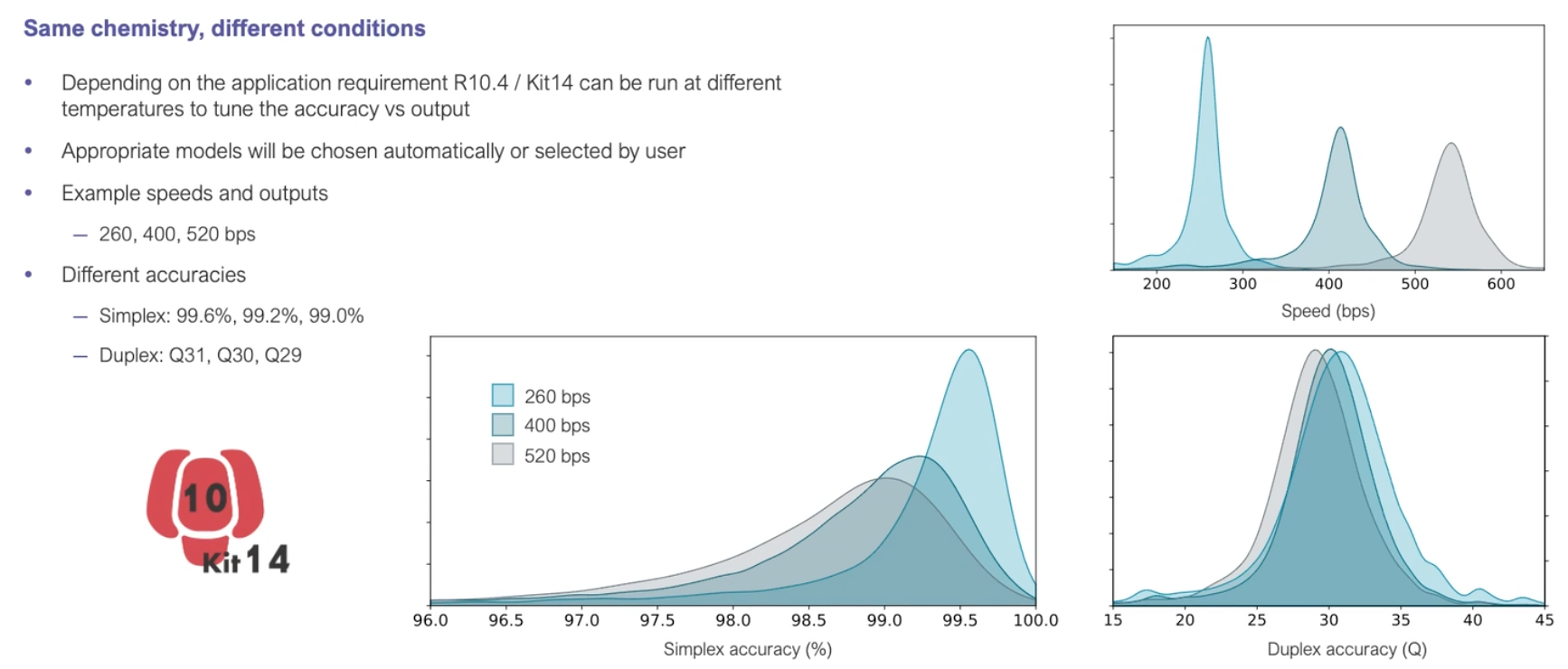
What aren’t they telling us about those duplex reads…?
https://nanoporetech.com/resource-centre/london-calling-2022-update-oxford-nanopore-technologies
Coming… sometime - the Mk1D

https://nanoporetech.com/resource-centre/london-calling-2022-update-oxford-nanopore-technologies https://nanoporetech.com/products/minion-mk1d
More Nanopore: London Calling 2022

- Online conference held in May 2022
- Talk videos available online
- LOTS of really cool announcements and research applications
Key Points
ONT produce a range of popular sequencing platforms.
This technology is constantly advancing.
Data Transfer
Overview
Teaching: 10 min
Exercises: 10 minQuestions
How do I get data to and from NeSI?
Objectives
Understand how to transfer data with Globus.
Understand how to transfer data with
scp.Understand how to transfer data within JupyterHub.
Up until now all the data that we have used for the Otago Bioinformatics Spring School has been either already on NeSI or able to be downladed there from the internet. With bioinformatics workflows we usually start with needing to get our data from our computer to the computers being used for processing and analysis and then once that is done, we need to transfer it back.
There are three main ways to go about transferring data between your computer and NeSI. These are using scp on the commandline, through the upload/download option when running a Jupyter notebook session on NeSI, or through Globus.
Both scp and Jupyter methods are only really suitable for small data transfers (< 1 GB). For anything larger, it is recommended to use Globus.
Generating FASTQ data - basecalling
During the ONT sequencing run, the data generated on the flowcell are stored in FAST5 format. In order to perform further analysis, the FAST5 data needs to be converted to FASTQ (the de facto standard for storing sequence and associated base-level quality information).
Basecalling usually occurs while a sequencing run is underway, but there are some specific situations that will require you to “re-basecall” your data:
-
You didn’t perform any basecalling while sequencing - e.g., a MinION being run from a computer without a GPU (so real-time basecalling not possible)
-
You performed basecalling in FAST mode while sequencing, and now you would like to “re-basecall” using a more accurate model (e.g., super-high accuracy mode: SUP)
You may also have downloaded some FAST5 data (i.e., without basecalls), or have access to some older FAST5 data, and you would like re-basecall using a more recent version of ONT’s software.
In all of these cases, we need to get the FAST5 data onto a compute system where you have access to a GPU.
Today we will use NeSI for this.
The first step in the process is to transfer the FAST5 data to NeSI.
scp
From a terminal, scp can be used to transfer files to a remote computer.
The documentation for how to use scp specifically with NeSI can be found at https://support.nesi.org.nz/hc/en-gb/articles/360000578455-File-Transfer-with-SCP. There are some configuration steps that need to be done to make this a bit easier.
The general format is scp <local file to transfer> mahuika:<path to copy file to> to send a file to NeSI, and scp mahuika:<path to file> <local path to copy to> to copy a file from NeSI to your machine.
Jupyter
Jupyter can be used to both upload and download data, however similar to scp it’s not ideal for sending large quantities of data.
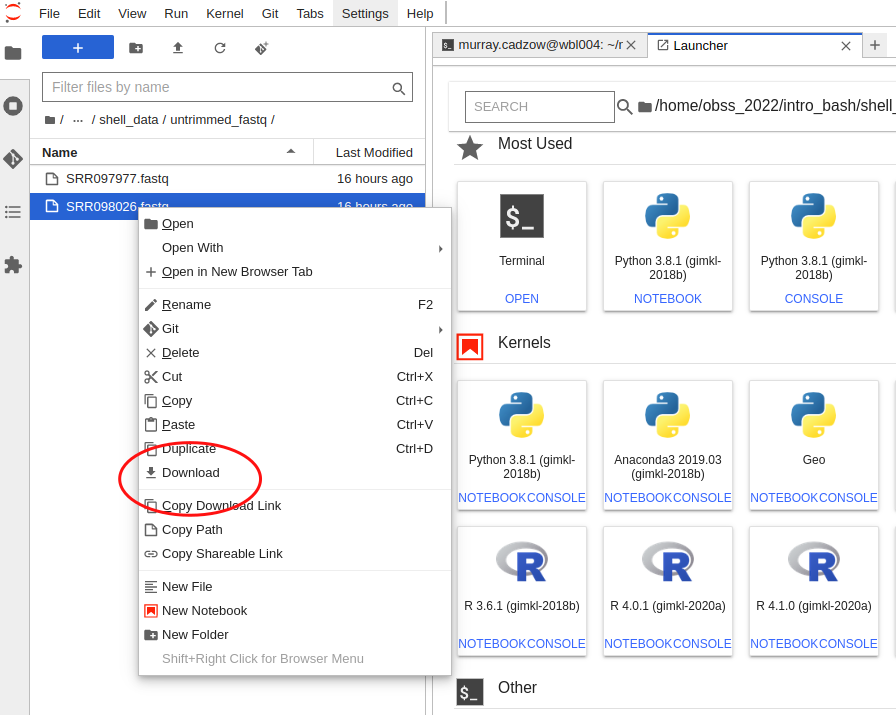
Downloading data
To download a file using Jupyter, you will need to use the explorer panel and navigate to the file there. Then, right-click on th file and a menu will pop up. Select “Download” from this menu and your browser will start downloading it.
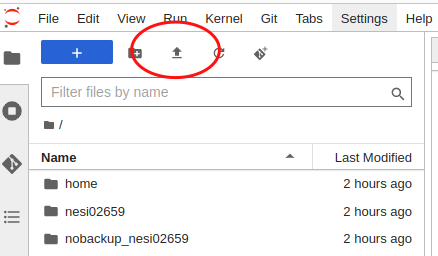
Uploading data
Similar to downloading through Juptyer, you first need to use the explorer panel to navigate to the directory where you would like the file to be uploaded to. From there click the upload button in the top left corner and select the file you wish to upload from your computer.
Globus
Globus is specialised software that can handle the transfer of large amounts of data between two ‘endpoints’. As part of the transfer it can automatically perform the file integrity checks to ensure the file was correctly transferred. The documentation for using Globus with NeSI is found at https://support.nesi.org.nz/hc/en-gb/articles/4405623380751-Data-Transfer-using-Globus-V5.
NeSI provides an endpoint, and you need to provide the second - this might be the globus software running on a different server, or the globus software running on your computer.
Globus Personal Connect
If you need to transfer data from your own machine, you can create your own endpoint by installing and running the Globus Personal Connect software avaiable from https://www.globus.org/globus-connect-personal.
Key Points
There are multiple methods to transfer data to and from NeSI.
The method of choice depends on the size of the data.
Nanopore - Basecalling
Overview
Teaching: 10 min
Exercises: 10 minQuestions
How do you perform offline (i.e., non-live) basecalling for ONT data?
Objectives
Understand FASTQ format.
Use guppy_basecaller on NeSI to generate FASTQ data from FAST5 output files.
Data generation
During the sequencing run, the electric trace (“squiggle”) data are stored in .fast5 files, which utilse the HDF5 file format:
“Hierarchical Data Format (HDF) is a set of file formats (HDF4, HDF5) designed to store and organize large amounts of data. Originally developed at the National Center for Supercomputing Applications, it is supported by The HDF Group, a non-profit corporation whose mission is to ensure continued development of HDF5 technologies and the continued accessibility of data stored in HDF.”
https://www.neonscience.org/resources/learning-hub/tutorials/about-hdf5
-
A single ONT flowcell produces a large amount of data - very roughly, 1Gbp of sequence data requires 1GB of storage (e.g., as gzipped fastq), but to generate 1Gbp of sequence requires 10GB of electrical trace data, so potentially up to 500GB of files for a 72 hour MinION run (a 30Gbp run will typically generate 300-400GB of
.fast5files). -
As the data are generated, they are saved as
.fast5files in either thefast5_passorfast5_failfolders, depending on whether reads passed or failed the quality assessment process, based on the criteria that were specified when setting up the run in MinKNOW. -
Each
.fast5file contains squiggle data for 1000 reads, so MANY files are created in a single sequencing run (e.g., a run generating five millions reads will produce 5000.fast5files, which are split across thefast5_passandfast5_failfolders). -
If realtime basecalling is performed during the run, then there will also be the equivalent number of
.fastqfiles generated (1000 reads per file), split across thefastq_passandfastq_failfolders.
Basecalling workflow
In order to analyse the data from a sequencing run, the FAST5 “squiggle” data needs to be converted to base calls. The ONT software application “guppy” can be used to process FAST5 data into FASTQ format - this is the de facto standard for storage of sequence data and associated base-level quality scores.
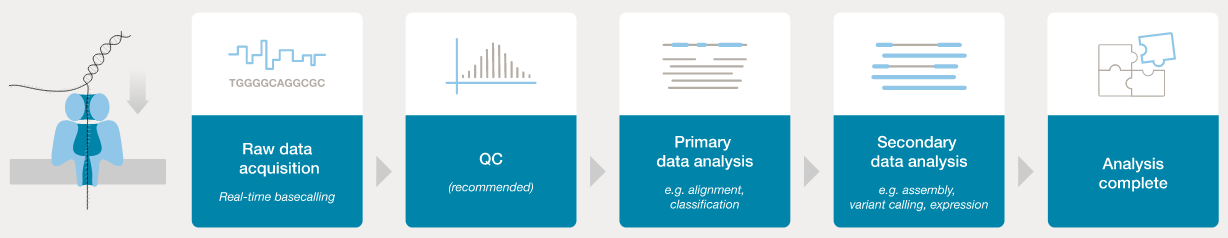
- ONT provides software (MinKNOW) for operating the MinION, and for generating the sequence data (e.g., the
guppybasecaller). - Once the raw FAST5 data have been converted to basecalls, we can use more familiar tools for quality assessment and analysis (e.g., FastQC).
https://nanoporetech.com/nanopore-sequencing-data-analysis
The current version of the Guppy software can be downloaded from the “Software Downloads” section of the Nanopore Community website:
https://community.nanoporetech.com/downloads
Note that you will need to create a login to access the website.
Today we will be using guppy_basecaller on the NeSI system to generate FASTQ data from a set of FAST5 files.
Recap: FASTQ data format
Assessing sequence quality: phred scores

Ewing B, Green P. (1998): Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8(3):186-194.

Can use ASCII to represent quality scores by adding 33 to the phred score and converting to ASCII.
- Quality score of 38 becomes 38+33=71: “G”
http://en.wikipedia.org/wiki/Phred_quality_score
- The FASTQ format allows the storage of both sequence and quality information for each read.
- This is a compact text-based format that has become the de facto standard for storing data from next generation sequencing experiments.
FASTQ format:

http://en.wikipedia.org/wiki/FASTQ_format
- Line 1: ‘@’ character followed by sequence identifier and optional description.
- Line 2: base calls.
- Line 3: ‘+’ character, optionally followed by the same sequence identifier (and description) again.
- Line 4: quality scores
E coli data set on NeSI
For this part of the workshop we are working at:
~/obss_2022/nanopore/ecoli-data/
Let’s change to that directory now:
cd ~/obss_2022/nanopore/ecoli-data/
FAST5 files from sequencing E. coli are available in the directory:
fast5_pass
ls -1 fast5_pass
ecoli_pass_0.fast5
ecoli_pass_10.fast5
ecoli_pass_11.fast5
ecoli_pass_12.fast5
ecoli_pass_13.fast5
ecoli_pass_14.fast5
ecoli_pass_15.fast5
ecoli_pass_1.fast5
ecoli_pass_2.fast5
ecoli_pass_3.fast5
ecoli_pass_4.fast5
ecoli_pass_5.fast5
ecoli_pass_6.fast5
ecoli_pass_7.fast5
ecoli_pass_8.fast5
ecoli_pass_9.fast5
Basecalling: guppy
guppyis a neural network based basecaller.- analyses the electrical trace data and predicts base
- it is GPU-aware, and can basecall in real time
- can also call base modifications (e.g., 5mC, 6mA)
- high accuracy mode (slower) and super-high accuracy mode (even slower) can improve basecalls post-sequencing
- The software is provided by ONT, and the source code is available via a developer agreement.
- Output is the standard “FASTQ” format for sequence data.
On NeSI:
module load ont-guppy-gpu/6.2.1
guppy_basecaller --help
: Guppy Basecalling Software, (C) Oxford Nanopore Technologies plc. Version 6.2.1+6588110, minimap2 version 2.22-r1101
Use of this software is permitted solely under the terms of the end user license agreement (EULA).By running, copying or accessing this software, you are demonstrating your acceptance of the EULA.
The EULA may be found in /scale_wlg_persistent/filesets/opt_nesi/CS400_centos7_bdw/ont-guppy-gpu/6.2.1/bin
Usage:
With config file:
guppy_basecaller -i <input path> -s <save path> -c <config file> [options]
With flowcell and kit name:
guppy_basecaller -i <input path> -s <save path> --flowcell <flowcell name>
--kit <kit name>
List supported flowcells and kits:
guppy_basecaller --print_workflows
Use GPU for basecalling:
guppy_basecaller -i <input path> -s <save path> -c <config file>
--device <cuda device name> [options]
Command line parameters:
[ TRUNCATED - this goes on for a long time... ]
Using guppy on NeSI
The guppy_basecaller software can use GPUs to speed up the basecalling process.
On NeSI, we can access the A100 GPUs by submitting a job to the cluster using a slurm script.
The slurm script we will use can be found at:
scripts/guppy_fastmodel.sl
Let’s have a look at the content:
more scripts/guppy_fastmodel.sl
#!/bin/bash -e
#SBATCH --job-name guppy_basecalling
#SBATCH --time 00:10:00
#SBATCH --mem 4G
#SBATCH --cpus-per-task 2
#SBATCH --output guppy_%j.out
#SBATCH --account nesi02659
#SBATCH --gpus-per-node A100-1g.5gb:1
module purge
module load ont-guppy-gpu/6.2.1
guppy_basecaller -i fast5_pass -s fastq_fastmodel \
--config /opt/nesi/CS400_centos7_bdw/ont-guppy-gpu/6.2.1/data/dna_r9.4.1_450bps_fast.cfg \
--device auto --recursive --records_per_fastq 4000 --min_qscore 7 --compress_fastq
- The
SBATCHcommands are providing information to theslurmjob scheduler: job name, maximum run time, memory allocation etc - The
modulecommands are making sure the necessary modules are available to the script (here we are specifying version 6.2.1 ofont-guppy-gpu- the GPU-enabled version of ONT’s guppy software). - The
guppy_basecallercommand is what is doing the work (i.e., converted the .fast5 data to .fastq).
Let’s have a closer look at the guppy_basecaller command:
guppy_basecaller \
-i fast5_pass_subset \
-s fastq_fastmodel \
--config /opt/nesi/CS400_centos7_bdw/ont-guppy-gpu/6.2.1/data/dna_r9.4.1_450bps_fast.cfg \
--device auto \
--recursive \
--records_per_fastq 4000 \
--min_qscore 7 \
--compress_fastq
Options used:
-i fast5_pass_subset: where to find the fast5 files (note the we are assuming that the script in run from the directory:~/obss_2022/nanopore/)-s fastq_fastmodel: the name of the folder where the .fastq files will be written to.-config /opt/nesi/CS400_centos7_bdw/ont-guppy-gpu/6.2.1/data/dna_r9.4.1_450bps_fast.cfg: this specifies the model that is being used to perform the basecalling. In this case we are using the “fast” model (rather than high-accuracy (hac) or super-high accuracy (sup)) to save time, anddnadenotes that we sequenced… DNA.r9.4.1refers to the flowcell version that was used to generate the data, and 450bps is the speed at which the DNA passes through the pore (450 bases per second).--device auto: this refers to which GPU device should be used (here we let the software selectautomatically, but it can sometimes be useful to select specific GPU devices).--recursive: search recursively for.fast5files (actually all our.fast5files are in the folder, so this isn’t needed here).--records_per_fastq 4000: number of reads to include per.fastqfile.--min_qscore 7: minimum average base quality score to considered a read to have “passed” QC.--compress_fastq: create compressed (.fastq.gz) files.
To submit the script, we use the sbatch command, and run it from the ~/obss_2022/nanopore/ecoli-data directory. You can check
if you are in that directory with pwd. If not: cd ~/obss_2022/nanopore/ecoli-data.
To run the script:
sbatch scripts/guppy_fastmodel.sl
Once the script has been submitted, you can check to make sure it is running via:
# This shows all your current jobs
squeue --me
JOBID USER ACCOUNT NAME CPUS MIN_MEM PARTITI START_TIME TIME_LEFT STATE NODELIST(REASON)
31430356 michael. nesi02659 spawner-jupy 2 4G infill 2022-11-20T0 1:23:52 RUNNING wbl004
31431766 michael. nesi02659 guppy_baseca 2 4G gpu 2022-11-20T0 6:28 RUNNING wbl001
It will also write progress to a log file that contains the job ID (represented by XXXXXXX in the code below).
You can check the file name using ls -ltr (list file details, in reverse time order).
Using the tail -f command, we can watch the progress (use control-c to exit):
ls -ltr
tail -f guppy_XXXXXXX.out
Example of output:
Use of this software is permitted solely under the terms of the end user license agreement (EULA).By running, copying or accessing this software, you are demonstrating your acceptance of the EULA.
The EULA may be found in /scale_wlg_persistent/filesets/opt_nesi/CS400_centos7_bdw/ont-guppy-gpu/6.2.1/bin
Found 16 fast5 files to process.
Init time: 847 ms
0% 10 20 30 40 50 60 70 80 90 100%
|----|----|----|----|----|----|----|----|----|----|
****************************
Use “Control-c” to exit (it’s okay, it won’t kill the job).
The job should take around 6 minutes to run.
Once the job has completed successfully, the .fastq files can be found at fastq_fastmodel:
ls -1 fastq_fastmodel
Note that pass and fail subfolders have been created.
ls -l fastq_fastmodel/pass/
ls -l fastq_fastmodel/fail/
Note that both directories (pass and fail) contain the same number of files, and they have exactly the same names. BUT, their content is different: the .fastq.gz files in the pass folder contain data for the reads that have passed QC (average base quality above threshold), and the fail folder contains those that failed. The files in the pass folder should be (hopefully!) much larger those their counterparts in the fail folder.
ls -lh fastq_fastmodel/pass/fastq_runid_15ca037c8ec4904bf5408d8ae5a1abecb8458aee_0_0.fastq.gz
-rw-rw----+ 1 michael.black nesi02659 5.7M Nov 20 00:58 fastq_fastmodel/pass/fastq_runid_15ca037c8ec4904bf5408d8ae5a1abecb8458aee_0_0.fastq.gz
ls -lh fastq_fastmodel/fail/fastq_runid_15ca037c8ec4904bf5408d8ae5a1abecb8458aee_0_0.fastq.gz
-rw-rw----+ 1 michael.black nesi02659 672K Nov 20 00:58 fastq_fastmodel/fail/fastq_runid_15ca037c8ec4904bf5408d8ae5a1abecb8458aee_0_0.fastq.gz
Here the first .fastq.gz file (in the pass folder) contains 5.7M of data, whereas the one in the fail folder only contains 672K (i.e., it is about 1/12 of the size).
Key Points
FAST5 data can be converted to FASTQ via the guppy_basecaller software.
guppy_basecaller has different models (FAST, HAC and SUP) that provide increasing basecall accuracy (and take increasing amounts of computation).
Once bascalling has been done, standard tasks such as QA/QC, mapping and variant calling can be performed.
Nanopore: quality assessment
Overview
Teaching: 10 min
Exercises: 10 minQuestions
How can I perform quality assessment of ONT data?
Objectives
Assess the quality of reads.
Generate summary metrics.
Software for quality assessment
In this part of the module, we’ll look at software applications that can be used to assess the quality of data from ONT sequencing runs. We’ll be focussing on two pieces of software: FastQC and NanoPlot.
- FastQC (you’ve already seen this): generic tool for assessing the quality of next generation sequencing data.
- NanoPlot: quality assessment software specifically built for ONT data.
NanoPlot
Let’s use the NanoPlot software to assess the quality of our .fastq data. We can run NanoPlot interactively (i.e., we don’t need to submit it as a slurm job), and it wil produce an HTML-based report that can be viewed in a browser.
Load the NanoPlot module:
module load NanoPlot
Run NanoPlot on passed fastq data (i.e., just the files in the pass folder):
NanoPlot -o nanoplot_fastmodel \
-p ecoli_fastmodel_subset_ \
--fastq fastq_fastmodel/pass/fastq_runid_*.gz
Command syntax:
NanoPlot: run the NanoPlot command (note that the capital N and P are required).-o nanoplot_fastmodel: folder for output (-o) to be written.-p ecoli_fastmodel_subset_: prefix (-p) to be appended to start of each filename in the output folder.--fastq fastq_fastmodel/pass/fastq_runid_*.gz: the.fastq.gzfiles to process with NanoPlot.
Let’s check what output was generated:
ls -1 nanoplot_fastmodel/
ecoli_fastmodel_subset_LengthvsQualityScatterPlot_dot.html
ecoli_fastmodel_subset_LengthvsQualityScatterPlot_kde.html
ecoli_fastmodel_subset_NanoPlot_20221120_2127.log
ecoli_fastmodel_subset_NanoPlot-report.html
ecoli_fastmodel_subset_NanoStats.txt
ecoli_fastmodel_subset_Non_weightedHistogramReadlength.html
ecoli_fastmodel_subset_Non_weightedLogTransformed_HistogramReadlength.html
ecoli_fastmodel_subset_WeightedHistogramReadlength.html
ecoli_fastmodel_subset_WeightedLogTransformed_HistogramReadlength.html
ecoli_fastmodel_subset_Yield_By_Length.html
The file that we are interested in is the HTML report: ecoli_fastmodel_subset_NanoPlot-report.html
To view this in your browser:
- Use the file browser (left panel of Jupyter window) to navigate to the
nanoplot_fastmodelfolder. - Control-click (Mac) or right-click (Windows/Linux) on the “ecoli_fastmodel_subset_NanoPlot-report.html” file and choose “Open in New Browser Tab”.
- The report should now be displayed in a new tab in your browser.
FastQC
To run the FastQC application on our FASTQ data, we need to create a single .fastq.gz file containing all our data (remember the data are currently spread across multiple .fastq.gz files in the fastq_fastmodel folder).
# Make a single .fastq.gz file:
cat fastq_fastmodel/pass/*.gz > ecoli-pass.fastq.gz
# Use zcat to decompress the data on-the-fly and stream it to wc to count the number of lines in the file
zcat ecoli-pass.fastq.gz | wc -l
# Use bc to calculate the number of reads (remember: 4 lines per read):
echo 56448/4 | bc
Load the FastQC module:
module load FastQC
Make a folder for the FastQC output to written to:
mkdir fastqc_fastmodel
Run FastQC on our data:
# NB: I get a java memory error if I don't use two threads
# FastQC allocates 250Mb per thread. Can't remember how
# to specify more memory for java...
fastqc -t 2 -o fastqc_fastmodel ecoli-pass.fastq.gz
Command syntax:
fastqc: run thefastqccommand-t 2: use two cpus (see my note about memory usage above) - the-tis for “threads”.-o fastqc_fastmodel: specify output folder.ecoli-pass.fastq.gz: FASTQ data to analyse.
Check the output:
ls -1 fastqc_fastmodel
ecoli-pass_fastqc.html
ecoli-pass_fastqc.zip
As we did with the NanoPlot output, we can view the HTML report in the browser:
- Use the file browser (left panel of Jupyter window) to navigate to the
fastqc_fastmodelfolder. - Control-click (Mac) or right-click (Windows/Linux) on the “ecoli-pass_fastqc.html” file and choose “Open in New Browser Tab”.
- The report should now be displayed in a new tab in your browser.
Bioawk
FastQC and NanoPlot are great for producing pre-formatted reports that summarise read/run quality.
But what if you want to ask some more specific questions about your data?
Bioawk is an extension to the Unix awk command which allows use to query specific biological data types - here we will use it to ask some specific questions about our FASTQ data.
Load the bioawk module.
module load bioawk
Extract read name and length:
bioawk -c fastx '{print $name,length($seq)}' ecoli-pass.fastq.gz | head
04670db1-f5f2-41f6-a6bd-92f43f6d3968 4403
0d91f59f-9fcd-4935-b062-8f05258816c5 12428
0c6a1ddc-1098-472e-a219-6d5c336e9e1b 2996
0e1306e1-785a-4307-8bbb-a5095f022306 4249
101a659f-fb10-4a22-8174-3f659c6ce462 772
106a9dc6-5e05-4c93-8ff1-569dc97426ca 6377
0f90ec4c-6fc9-4592-8754-bf4fb800d115 2452
11fffe54-669b-4fbf-be13-2236507aa8c9 8762
13f7a628-acda-4f6e-927d-651cb62adf51 14868
14a422e7-c54b-4a1a-9a64-a68ce69e4c5c 7185
Number of reads:
bioawk -c fastx '{print $name,length($seq)}' ecoli-pass.fastq.gz | wc -l
14112
NB: this SHOULD match what we calculated above.
Total number of bases in first ten reads (I’ve stepped through the process to illustrate each component of the command):
bioawk -c fastx '{print length($seq)}' ecoli-pass.fastq.gz | head
4403
12428
2996
4249
772
6377
2452
8762
14868
7185
bioawk -c fastx '{print length($seq)}' ecoli-pass.fastq.gz | head | paste -sd+
4403+12428+2996+4249+772+6377+2452+8762+14868+7185
bioawk -c fastx '{print length($seq)}' ecoli-pass.fastq.gz | head | paste -sd+ | bc
64492
So the first 10 reads comprise 64,492 bases.
Total number of bases in all reads
bioawk -c fastx '{print length($seq)}' ecoli-pass.fastq.gz | paste -sd+ | bc
144479297
In total, the 14112 reads in our data set comprise 144,479,297 bases (144Mbp).
Number of reads longer than 10000 bases
First create “yes/no” (1/0) information about whether each read has a length greater than 10,000 bases:
bioawk -c fastx '{print (length($seq) > 10000)}' ecoli-pass.fastq.gz | head
0
1
0
0
0
0
0
0
1
0
Now sum this up to find the total number of reads satisfying this condition:
bioawk -c fastx '{print (length($seq) > 5000)}' ecoli-pass.fastq.gz | paste -sd+ | bc
8700
So, of our 14,112 reads, 8700 of them are longer than 10,000 bases.
Key Points
FastQC and NanoPlot provide pre-formatted reports on run characteristics and read quality.
Bioawk can be used to generate summary metrics and ask specific questions about the read data.
Nanopore - Variant Calling
Overview
Teaching: 10 min
Exercises: 10 minQuestions
How can I use nanopore data for variant calling?
Objectives
Align reads from long-read data.
Perform variant calling using nanopore data.
Sequence alignment
- If a “reference” genome exists for the organism you are sequencing, reads can be “aligned” to the reference.
- This involves finding the place in the reference genome that each read matches to.
- Due to high sequence similarity within members of the same species, most reads should map to the reference.
Tools for generating alignments
- There are MANY software packages available for aligning data from next generation sequencing experiments.
- For short-read data e.g., Illumina sequencing), the two “original” short read aligners were:
- BWA: http://bio-bwa.sourceforge.net
- Bowtie: http://bowtie-bio.sourceforge.net
- Over the years, MANY more “aligners” have been developed.
- For long-read data, ONT provides the Minimap2 aligner (but there are also a number of other options).
Alignment with minimap2 on NeSI
Load the minimap2 module:
module load minimap2
Check that we’ve got a reference genome:
ls genome
e-coli-k12-MG1655.fasta
Load the SAMtools module:
module load SAMtools
Make a folder for our aligned data:
mkdir bam-files
Use minimap2 followed by samtools to generate aligned data in .bam format:
minimap2 -ax map-ont genome/e-coli-k12-MG1655.fasta \
fastq_fastmodel/pass/*.gz | \
samtools sort -o bam-files/ecoli-pass-aligned-sort.bam
Index the bam file:
samtools index bam-files/ecoli-pass-aligned-sort.bam
Generate basic mapping information using samtools:
samtools coverage bam-files/ecoli-pass-aligned-sort.bam
#rname startpos endpos numreads covbases coverage meandepth meanbaseq meanmapq
U00096.3 1 4641652 14273 4641652 100 28.93 17.3 59.8
Variant calling
NB - code below is taken from the “Variant Calling Workflow” section of the Genomic Data Carpentry course:
https://datacarpentry.org/wrangling-genomics/04-variant_calling/index.html
Load the BCFtools module:
module load BCFtools
Make a folder for the output data:
mkdir bcf-files
Generate a “pileup” file to enable variant calling (might take a little bit of time):
bcftools mpileup -B -Q5 --max-BQ 30 -I \
-o bcf-files/ecoli-pass.bcf \
-f genome/e-coli-k12-MG1655.fasta \
bam-files/ecoli-pass-aligned-sort.bam
Make a folder to hold vcf (variant call format) files:
mkdir vcf-files
Use bcftools to call variants in the e. coli data:
bcftools call --ploidy 1 -m -v \
-o vcf-files/ecoli-variants.vcf \
bcf-files/ecoli-pass.bcf
Use vcfutils.pl to filter the variants (in this case, everything gets kept):
vcfutils.pl varFilter vcf-files/ecoli-variants.vcf > vcf-files/ecoli-final-variants.vcf
more vcf-files/ecoli-final-variants.vcf
Key Points
Long-read based algorithms (e.g., minimap2) need to be used when aligning long read data.
Post-alignment, standard software tools (e.g., samtools, bcftools, etc) can be used to perform variant calling.
Nanopore - Adaptive Sampling
Overview
Teaching: 10 min
Exercises: 10 minQuestions
How can nanopore be used for sequencing only what I want?
Objectives
Explain how adaptive sampling works
Let’s learn about Adaptive Sampling…
Mik will now talk at you. :)
Now let’s take a look at some data
The folder adaptive-sampling contains data from a SIMULATED adaptive sampling run using David Eccles’ yoghurt data:
https://zenodo.org/record/2548026#.YLifQOuxXOQ
What did I do?
The bulk fast5 file was used to simulate a MinION run. Instead of just playing through the run file which would essentially just regenerate the exact same data that the run produced initially), I used MinKNOW’s built-in adaptive sampling functionality.
-
Specified S. thermophilus as the target genome (1.8Mbp).
-
Provided a bed file that listed 100Kbp regions, interspersed with 100Kbp gaps:
NZ_LS974444.1 0 99999
NZ_LS974444.1 200000 299999
NZ_LS974444.1 400001 500000
NZ_LS974444.1 600000 699999
NZ_LS974444.1 800000 899999
NZ_LS974444.1 1000000 1099999
NZ_LS974444.1 1200000 1299999
NZ_LS974444.1 1400000 1499999
NZ_LS974444.1 1600000 1699999
Note that NZ_LS974444.1 is the name of the single chromosome in the fasta file.
Also note that the bed format starts counting at 0 (i.e., the first base of a chromosome is considered “base 0”).
- Specified that we wanted to enrich for these regions.
Some caveats:
-
We are pretending to do an adaptive sampling run, using data generated from a real (non-AS) run, so when a pore is unblocked (i.e., a read mapping to a region that we don’t want to enrich for) in our simulation, we stop accumulating data for that read, but the pore doesn’t then become available for sequencing another read (because in the REAL run, the pore continued to sequence that read).
-
This sample had a LOT of short sequences, so in many cases, the read had been fully sequenced before the software had a chance to map it to the genome and determine whether or not to continue sequencing.
Distribution of read lengths
Here is the full distribution of read lengths without adaptive sampling turned on (i.e., I also ran a simulation without adaptive sampling) - there are LOTS of reads that less than 1000bp long:
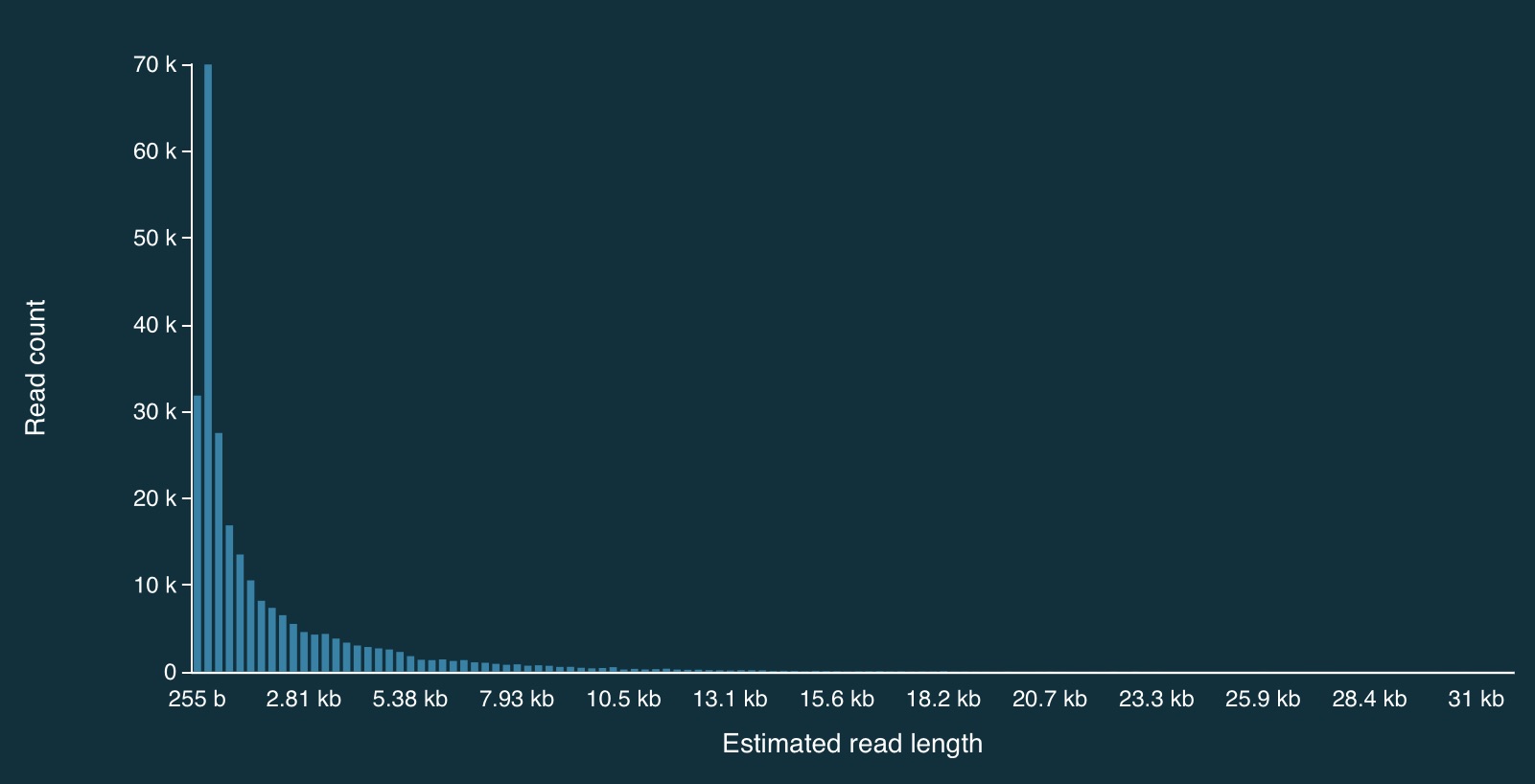
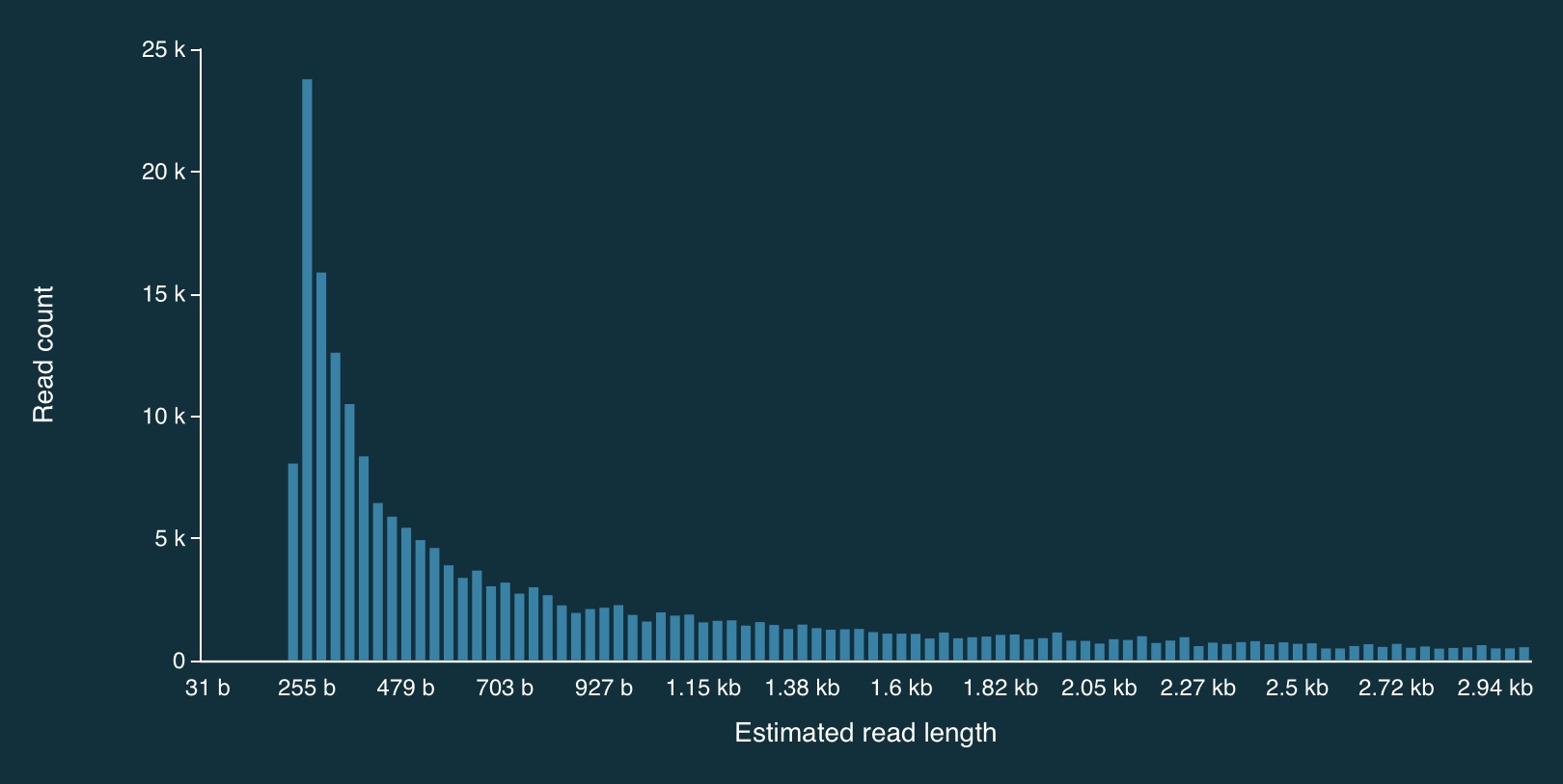
Here is a zoomed in view:
Adaptive sampling results
Despite the caveats, it is fairly clear that the adaptive sampling algorithm worked (or at least that it did something). In the plots below, the green bars represent reads that were not unblocked (i.e., the AS algorithm either: (1) decided the read mapped to a region in our bed file and continued sequencing it until the end, or (2) completed sequenced before a decision had been made), while the purple bars represent reads that were unblocked (i.e., the read was mapped in real-time, and the AS algorithm decided that it did not come from a region in our bed file, so ejected the read from the pore).
Here is the full distribution:
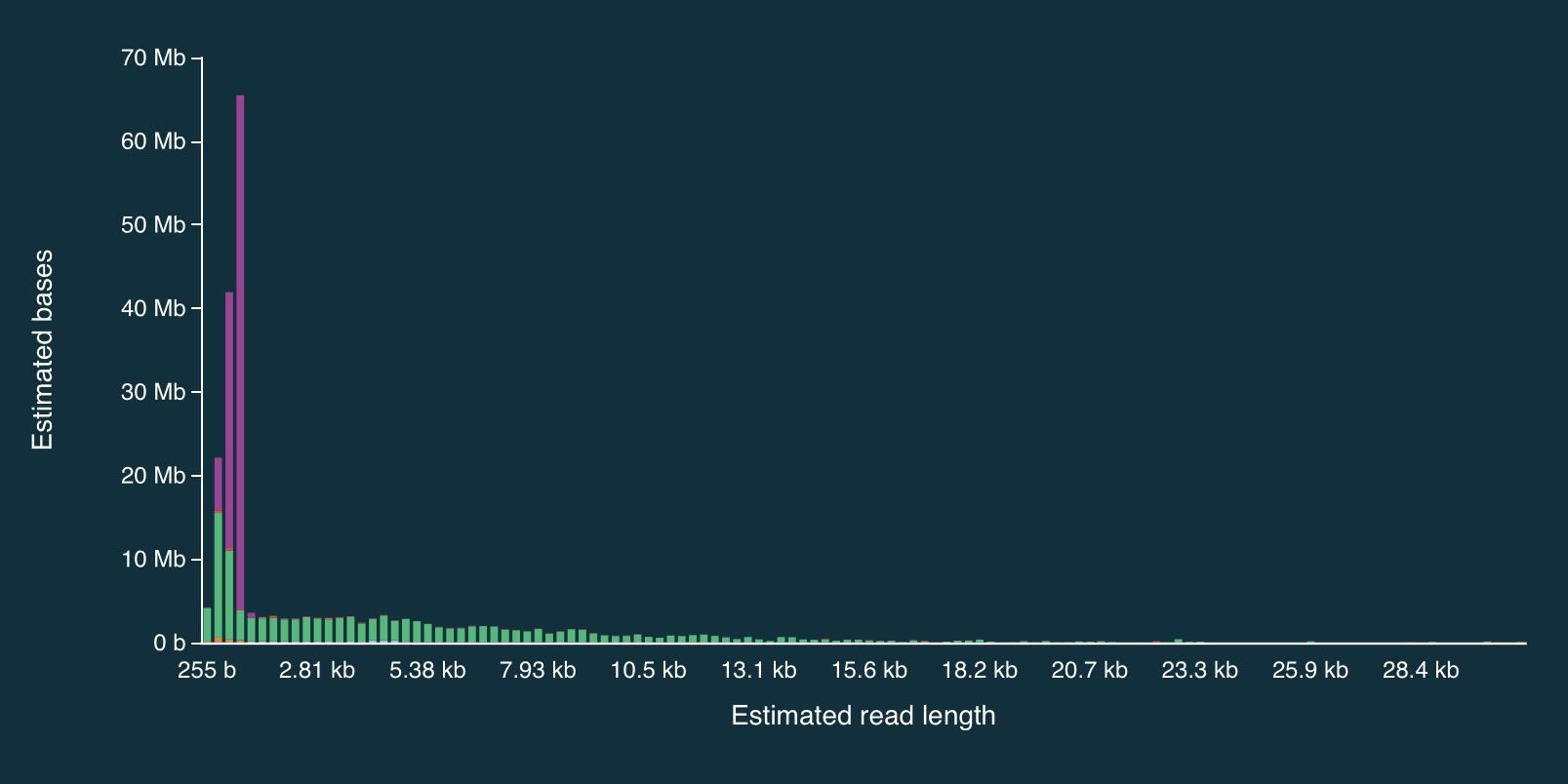
And here is a close up:
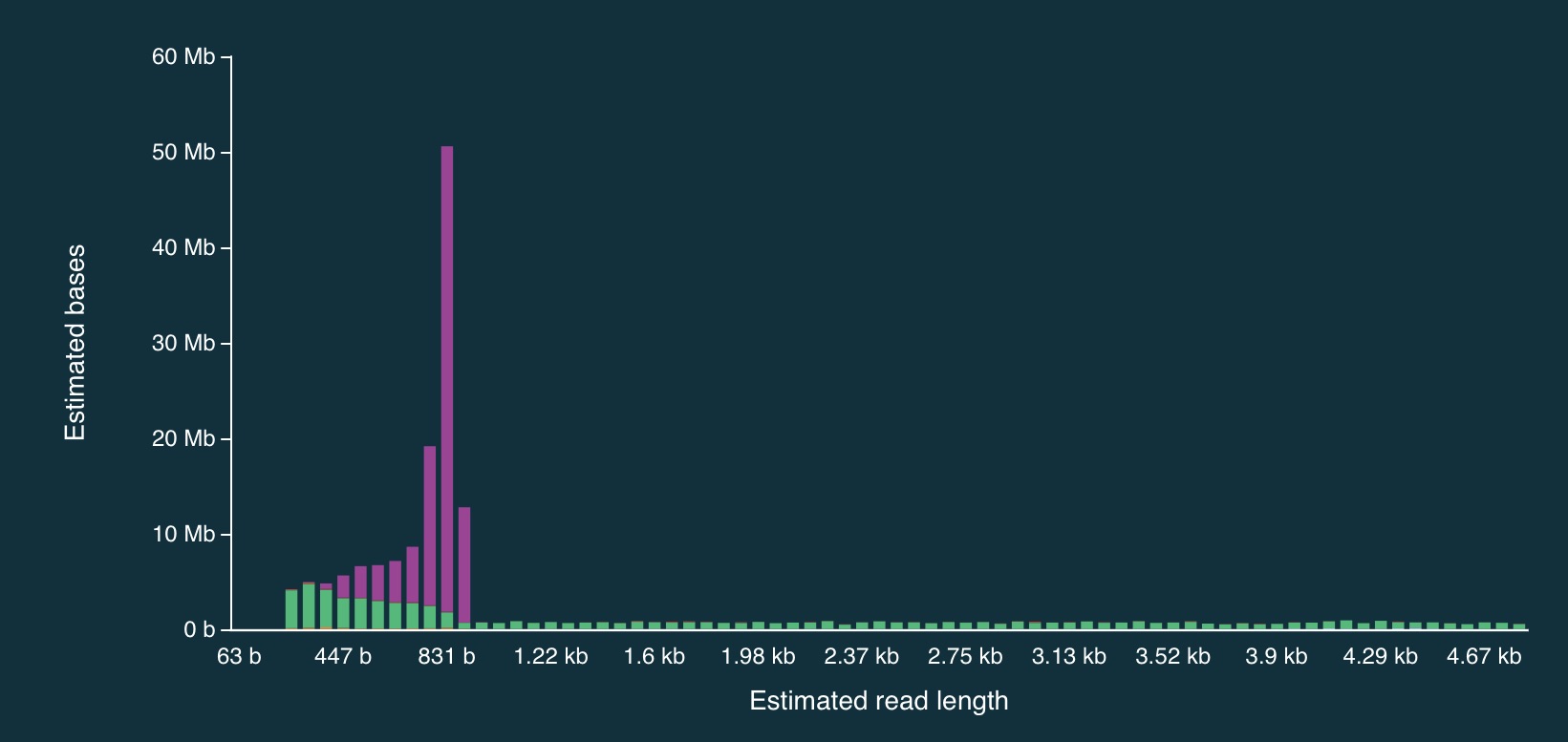
It is clear from the plots that the length of reads receiving an “unblock” signal is around 800bp, reflecting the time that the AS algorithm takes to make a decision about whether or not to reject a read.
Adaptive sampling details
When adaptive sampling is enabled, an additional output file is created, providing information about every read that was processed.
It is stored in the other_reports folder within the run directory, and is in CSV format. The naming convention is:
adaptive_sampling_XXX_YYY.csv
where XXC and YYY are flowcell and run identifiers (I think).
The format is:
batch_time,read_number,channel,num_samples,read_id,sequence_length,decision
1669157044.6601431,499,3,4008,bc65b9b4-bc05-45c4-a7b3-4c199577c8c9,350,unblock
1669157045.2247138,301,131,4000,1892041e-71a5-4121-9547-49122e247172,258,unblock
1669157045.2247138,438,42,4002,6be52363-5315-445c-bcd4-3d3b32a829dc,312,stop_receiving
1669157045.2247138,590,76,4001,e459305c-288d-47e7-9bbd-93224ee45074,264,unblock
1669157045.2247138,280,95,4005,a15f9aef-9b60-4f8f-bb74-c96b6f4fa16f,360,stop_receiving
1669157045.2247138,598,94,4001,8f615191-d1d4-42c5-a778-92c91411979b,357,unblock
1669157045.2247138,554,125,4003,8e628f9d-ae37-499e-b32e-7842bcf1539f,364,stop_receiving
1669157045.2247138,431,110,4003,12516158-8ed0-4bf2-bdff-f94171593996,378,unblock
1669157045.2247138,414,102,4008,1cad751a-ddde-4700-bb06-9dcf8974185a,313,unblock
Or slightly nicer (commas exchanged for tabs):
batch_time read_number channel num_samples read_id sequence_length decision
1669157044.6601431 499 3 4008 bc65b9b4-bc05-45c4-a7b3-4c199577c8c9 350 unblock
1669157045.2247138 301 131 4000 1892041e-71a5-4121-9547-49122e247172 258 unblock
1669157045.2247138 438 42 4002 6be52363-5315-445c-bcd4-3d3b32a829dc 312 stop_receiving
1669157045.2247138 590 76 4001 e459305c-288d-47e7-9bbd-93224ee45074 264 unblock
1669157045.2247138 280 95 4005 a15f9aef-9b60-4f8f-bb74-c96b6f4fa16f 360 stop_receiving
1669157045.2247138 598 94 4001 8f615191-d1d4-42c5-a778-92c91411979b 357 unblock
1669157045.2247138 554 125 4003 8e628f9d-ae37-499e-b32e-7842bcf1539f 364 stop_receiving
1669157045.2247138 431 110 4003 12516158-8ed0-4bf2-bdff-f94171593996 378 unblock
1669157045.2247138 414 102 4008 1cad751a-ddde-4700-bb06-9dcf8974185a 313 unblock
Processing adaptive sampling data
Once the data have been generated, they are processed in exactly the same way:
- Basecalling with
guppy_basecaller - QA/QC with FastQC and/or NanoPlot
- Alignment with
minimap2
I’ve done the above, and a bam file (wih an annoyingly long name) can be found at:
bam-files/yog-as-100kb-chunks-bed-SUP-pass-aligned-sort.bam
There are also a few additional things we can do to check how well the adaptive sampling has worked.
Filtering for read length
Because there are so many short reads (i.e., < 1000 bases) it is actually quite difficult to see the effect of adaptive sampling in this data set. To overcome this, we can remove these short reads from the data.
To do this, we can filter the bam file:
samtools view -h bam-files/yog-as-100kb-chunks-bed-SUP-pass-aligned-sort.bam | \
awk 'length($10) > 1000 || $1 ~ /^@/' | \
samtools view -bS - > bam-files/yog-as-100kb-chunks-bed-SUP-pass-aligned-sort-LONG-1KB.bam
And then index it:
samtools index bam-files/yog-as-100kb-chunks-bed-SUP-pass-aligned-sort-LONG-1KB.bam
Now we have a new bam file that only contains aligned reads longer than 1000bp.
Checking the read depth of the selected regions
We can use samtools bedcov to calculate how much sequence data was generated for specific regions of the genome.
For this, we need a bed file. This one breaks the 1.8Mbp S. thermophilus genome into 100Kbp chunks (admitedly, I missed a little bit of the end - after 1,800,000bp):
NZ_LS974444.1 0 99999
NZ_LS974444.1 100000 199999
NZ_LS974444.1 200000 299999
NZ_LS974444.1 300000 399999
NZ_LS974444.1 400000 499999
NZ_LS974444.1 500000 599999
NZ_LS974444.1 600000 699999
NZ_LS974444.1 700000 799999
NZ_LS974444.1 800000 899999
NZ_LS974444.1 900000 999999
NZ_LS974444.1 1000000 1099999
NZ_LS974444.1 1100000 1199999
NZ_LS974444.1 1200000 1299999
NZ_LS974444.1 1300000 1399999
NZ_LS974444.1 1400000 1499999
NZ_LS974444.1 1500000 1599999
NZ_LS974444.1 1600000 1699999
NZ_LS974444.1 1700000 1799999
Aside: how can we generate a file like that?
Let’s build one with bash!
First, let’s write a loop that counts from 1 to 10 in steps of 2:
for i in {1..10..2}
do
echo $i
done
1
3
5
7
9
Now lets do 0 to 1,799,999, in steps of 100,000:
for i in {0..1799999..100000}
do
echo $i
done
0
100000
200000
300000
400000
500000
600000
700000
800000
900000
1000000
1100000
1200000
1300000
1400000
1500000
1600000
1700000
Add the chromosome name, and the end position to each line (and make sure to separate with tabs):
for i in {0..1799999..100000}
do
start=$i
end=`echo $i+99999 | bc`
echo -e NZ_LS974444.1'\t'$start'\t'$end
done
NZ_LS974444.1 0 99999
NZ_LS974444.1 100000 199999
NZ_LS974444.1 200000 299999
NZ_LS974444.1 300000 399999
NZ_LS974444.1 400000 499999
NZ_LS974444.1 500000 599999
NZ_LS974444.1 600000 699999
NZ_LS974444.1 700000 799999
NZ_LS974444.1 800000 899999
NZ_LS974444.1 900000 999999
NZ_LS974444.1 1000000 1099999
NZ_LS974444.1 1100000 1199999
NZ_LS974444.1 1200000 1299999
NZ_LS974444.1 1300000 1399999
NZ_LS974444.1 1400000 1499999
NZ_LS974444.1 1500000 1599999
NZ_LS974444.1 1600000 1699999
NZ_LS974444.1 1700000 1799999
Using samtools bedcov
Once we’ve got a bed file, samtools bedcov can be used to calculate the amount of sequence generated for the
regions in the bed file.
I’ve generated a longer bed file, splitting the genome into 10Kb chunks. It can be found at:
bed-files/chunks-10k.bed
Now let’s use this file with samtools bedcov:
samtools bedcov bed-files/chunks-10k.bed \
bam-files/yog-as-100kb-chunks-bed-SUP-pass-aligned-sort-LONG-1KB.bam
First 20 rows:
NZ_LS974444.1 0 9999 1971063
NZ_LS974444.1 10000 19999 1311458
NZ_LS974444.1 20000 29999 1276564
NZ_LS974444.1 30000 39999 1744496
NZ_LS974444.1 40000 49999 1665578
NZ_LS974444.1 50000 59999 420445
NZ_LS974444.1 60000 69999 350856
NZ_LS974444.1 70000 79999 1138767
NZ_LS974444.1 80000 89999 1320115
NZ_LS974444.1 90000 99999 1393254
NZ_LS974444.1 100000 109999 415558
NZ_LS974444.1 110000 119999 131388
NZ_LS974444.1 120000 129999 189621
NZ_LS974444.1 130000 139999 156248
NZ_LS974444.1 140000 149999 154235
NZ_LS974444.1 150000 159999 127957
NZ_LS974444.1 160000 169999 105826
NZ_LS974444.1 170000 179999 105900
NZ_LS974444.1 180000 189999 61800
NZ_LS974444.1 190000 199999 315476
Write it to a text file:
mkdir coverage-files
samtools bedcov bed-files/chunks-10k.bed \
bam-files/yog-as-100kb-chunks-bed-SUP-pass-aligned-sort-LONG-1KB.bam > \
coverage-files/yog-as-100kb-chunks-10k-cov.txt
We’ll use this to make a plot of read depth across the genome in R.
But first…
Another aside: complementing a bed file
If you have a bed file containing genomic regions that have been used for adaptive sampling, it can be useful to create a second bed file that containing the complement of these regions (i.e., the regions of the genome that were not being selected via adaptive sampling). This can be used to check read depth at non-selected regions.
To do this, we first index the reference genome using samtools:
samtools faidx genome/streptococcus-thermophilus-strain-N4L.fa
This produces the file: streptococcus-thermophilus-strain-N4L.fa.fai:
more genome/streptococcus-thermophilus-strain-N4L.fa.fai
NZ_LS974444.1 1831756 66 70 71
We can use this to extract chromosome sizes (in this case, there is only one, but for multi-chromosome genomes - e.g., human - this is a really useful approach):
cut -f 1,2 genome/streptococcus-thermophilus-strain-N4L.fa.fai > genome/st-chrom-sizes.txt
The cut command splits lines of text into separate “fields” - by default, “tab” is used as the delimiter (others can be specified).
In this case, the code just takes the first two fields, which are the chromosome name and length.
Check what it produced:
more genome/st-chrom-sizes.txt
NZ_LS974444.1 1831756
This information can then be used to create the complement bed file:
module load BEDTools
bedtools complement -i bed-files/as-100kb-chunks.bed -g genome/st-chrom-sizes.txt > bed-files/as-100kb-chunks-complement.bed
more bed-files/as-100kb-chunks-complement.bed
NZ_LS974444.1 99999 200000
NZ_LS974444.1 299999 400000
NZ_LS974444.1 499999 600000
NZ_LS974444.1 699999 800000
NZ_LS974444.1 899999 1000000
NZ_LS974444.1 1099999 1200000
NZ_LS974444.1 1299999 1400000
NZ_LS974444.1 1499999 1600000
NZ_LS974444.1 1699999 1831756
Check (visually) that this is the complement of the bed file I used for adaptive sampling:
more bed-files/as-100kb-chunks.bed
NZ_LS974444.1 0 99999
NZ_LS974444.1 200000 299999
NZ_LS974444.1 400000 499999
NZ_LS974444.1 600000 699999
NZ_LS974444.1 800000 899999
NZ_LS974444.1 1000000 1099999
NZ_LS974444.1 1200000 1299999
NZ_LS974444.1 1400000 1499999
NZ_LS974444.1 1600000 1699999
We can use these files with samtools bedcov to generate read depth information separately for the
selected and non-selected regions (handy if you AS selection is somewhat complex - e.g., all exons, a specific set of genes etc).
Coverage info for selected regions:
samtools bedcov bed-files/as-100kb-chunks.bed \
bam-files/yog-as-100kb-chunks-bed-SUP-pass-aligned-sort-LONG-1KB.bam > \
coverage-files/yog-as-100kb-chunks-SELECTED-cov.txt
Coverage info for non-selected regions:
samtools bedcov bed-files/as-100kb-chunks-complement.bed \
bam-files/yog-as-100kb-chunks-bed-SUP-pass-aligned-sort-LONG-1KB.bam > \
coverage-files/yog-as-100kb-chunks-NONSELECTED-cov.txt
more coverage-files/yog-as-100kb-chunks-SELECTED-cov.txt
NZ_LS974444.1 0 99999 12593594
NZ_LS974444.1 200000 299999 14610340
NZ_LS974444.1 400000 499999 12060688
NZ_LS974444.1 600000 699999 14714058
NZ_LS974444.1 800000 899999 16128475
NZ_LS974444.1 1000000 1099999 15793991
NZ_LS974444.1 1200000 1299999 17615309
NZ_LS974444.1 1400000 1499999 13341368
NZ_LS974444.1 1600000 1699999 16061631
more coverage-files/yog-as-100kb-chunks-NONSELECTED-cov.txt
NZ_LS974444.1 99999 200000 1764394
NZ_LS974444.1 299999 400000 1738930
NZ_LS974444.1 499999 600000 1736471
NZ_LS974444.1 699999 800000 1602761
NZ_LS974444.1 899999 1000000 1739915
NZ_LS974444.1 1099999 1200000 2134827
NZ_LS974444.1 1299999 1400000 4654002
NZ_LS974444.1 1499999 1600000 3945477
NZ_LS974444.1 1699999 1831756 2277969
Some quick analysis in R
Choose “New Launcher” from the file menu, and start an RStudio session.
Genome-wide read-depth
Load the dplyr and ggplot2 packages:
Load the text file that we generated using samtools bedcov:
bedCov = read.table('coverage-files/yog-as-100kb-chunks-10k-cov.txt', header=FALSE, sep='\t')
Take a look at the data:
head(bedCov)
V1 V2 V3 V4
1 NZ_LS974444.1 0 9999 1971063
2 NZ_LS974444.1 10000 19999 1311458
3 NZ_LS974444.1 20000 29999 1276564
4 NZ_LS974444.1 30000 39999 1744496
5 NZ_LS974444.1 40000 49999 1665578
6 NZ_LS974444.1 50000 59999 420445
Add some column names:
names(bedCov) = c("CHROM", "START", "END", "BASES")
Add columns for region length (LENGTH) and then calculated average read depth (DEPTH):
bedCov = mutate(bedCov, LENGTH = END - START + 1)
bedCov = mutate(bedCov, DEPTH = BASES / LENGTH)
head(bedCov)
CHROM START END BASES LENGTH DEPTH
1 NZ_LS974444.1 0 9999 1971063 10000 197.1063
2 NZ_LS974444.1 10000 19999 1311458 10000 131.1458
3 NZ_LS974444.1 20000 29999 1276564 10000 127.6564
4 NZ_LS974444.1 30000 39999 1744496 10000 174.4496
5 NZ_LS974444.1 40000 49999 1665578 10000 166.5578
6 NZ_LS974444.1 50000 59999 420445 10000 42.0445
ggplot(bedCov, aes(x=START, y=DEPTH)) +
geom_line() +
ggtitle("Read depth across S. Thermophils genome (reads > 1Kbp)") +
xlab("Base position") + ylab("Read depth")
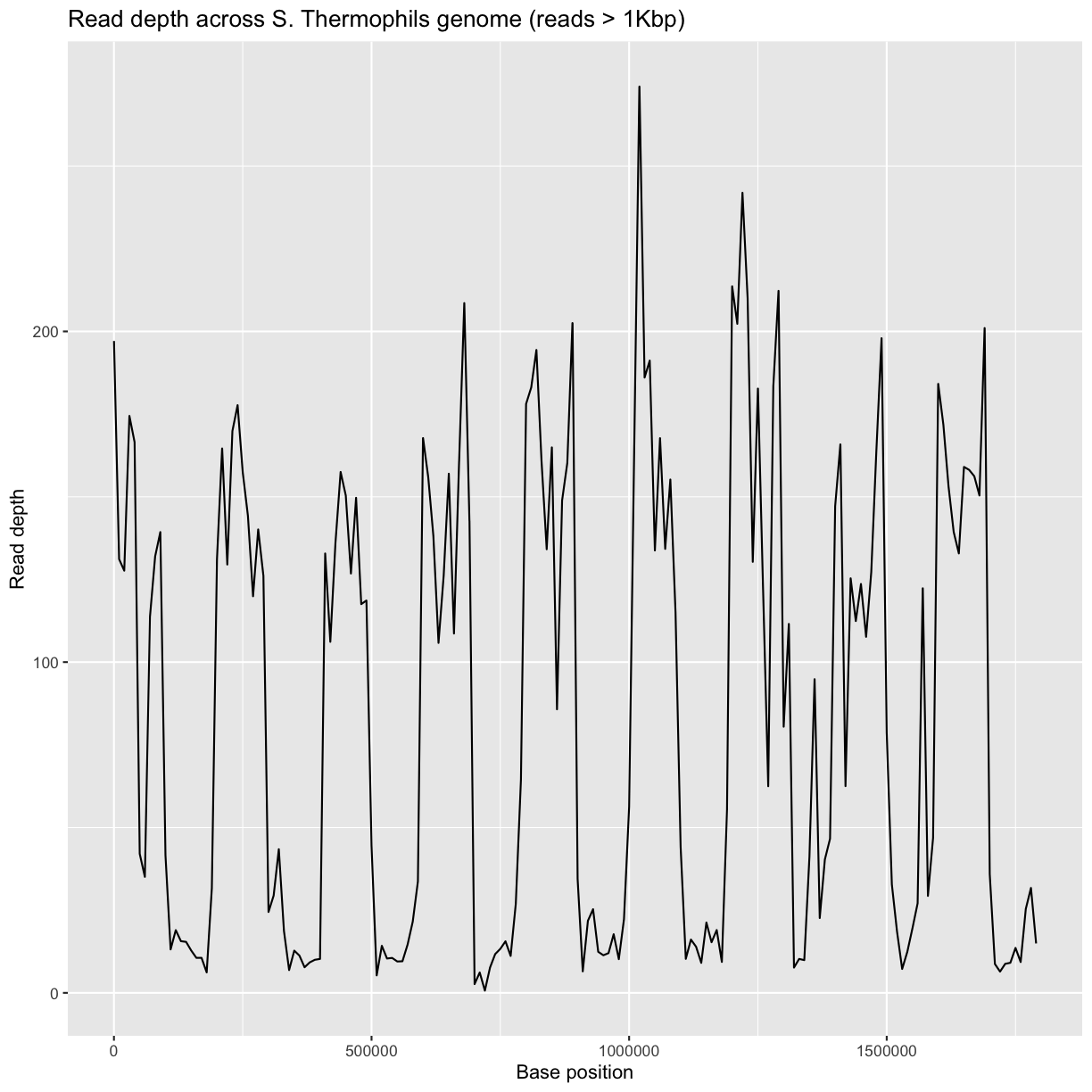
Average read-depth in selected and non-selected regions
bedCovSelected = read.table('coverage-files/yog-as-100kb-chunks-SELECTED-cov.txt', header=FALSE, sep='\t')
names(bedCovSelected) = c("CHROM", "START", "END", "BASES")
bedCovSelected = mutate(bedCovSelected, LENGTH = END - START + 1)
bedCovSelected = mutate(bedCovSelected, DEPTH = BASES / LENGTH)
Calculate average read depth for selected regions:
mean(bedCovSelected$DEPTH)
[1] 147.6883
Exercise: calculating read-depth
- Repeat the average read depth calculation for the non-selected regions.
- Was the average read depth different between the two regions?
Solution
bedCovNonSelected = read.table('coverage-files/yog-as-100kb-chunks-NONSELECTED-cov.txt', header=FALSE, sep='\t') names(bedCovNonSelected) = c("CHROM", "START", "END", "BASES") bedCovNonSelected = mutate(bedCovNonSelected, LENGTH = END - START + 1) bedCovNonSelected = mutate(bedCovNonSelected, DEPTH = BASES / LENGTH) mean(bedCovNonSelected$DEPTH)Average read depth for non-selected regions is: 23.38
This is A LOT less than the average read depth for the selected regions: 147.69
Key Points
Nanopore can determine if it should continue to sequence a read.