Nanopore - Basecalling
Overview
Teaching: 10 min
Exercises: 10 minQuestions
How do you perform offline (i.e., non-live) basecalling for ONT data?
Objectives
Understand FASTQ format.
Use guppy_basecaller on NeSI to generate FASTQ data from FAST5 output files.
Data generation
During the sequencing run, the electric trace (“squiggle”) data are stored in .fast5 files, which utilse the HDF5 file format:
“Hierarchical Data Format (HDF) is a set of file formats (HDF4, HDF5) designed to store and organize large amounts of data. Originally developed at the National Center for Supercomputing Applications, it is supported by The HDF Group, a non-profit corporation whose mission is to ensure continued development of HDF5 technologies and the continued accessibility of data stored in HDF.”
https://www.neonscience.org/resources/learning-hub/tutorials/about-hdf5
-
A single ONT flowcell produces a large amount of data - very roughly, 1Gbp of sequence data requires 1GB of storage (e.g., as gzipped fastq), but to generate 1Gbp of sequence requires 10GB of electrical trace data, so potentially up to 500GB of files for a 72 hour MinION run (a 30Gbp run will typically generate 300-400GB of
.fast5files). -
As the data are generated, they are saved as
.fast5files in either thefast5_passorfast5_failfolders, depending on whether reads passed or failed the quality assessment process, based on the criteria that were specified when setting up the run in MinKNOW. -
Each
.fast5file contains squiggle data for 1000 reads, so MANY files are created in a single sequencing run (e.g., a run generating five millions reads will produce 5000.fast5files, which are split across thefast5_passandfast5_failfolders). -
If realtime basecalling is performed during the run, then there will also be the equivalent number of
.fastqfiles generated (1000 reads per file), split across thefastq_passandfastq_failfolders.
Basecalling workflow
In order to analyse the data from a sequencing run, the FAST5 “squiggle” data needs to be converted to base calls. The ONT software application “guppy” can be used to process FAST5 data into FASTQ format - this is the de facto standard for storage of sequence data and associated base-level quality scores.
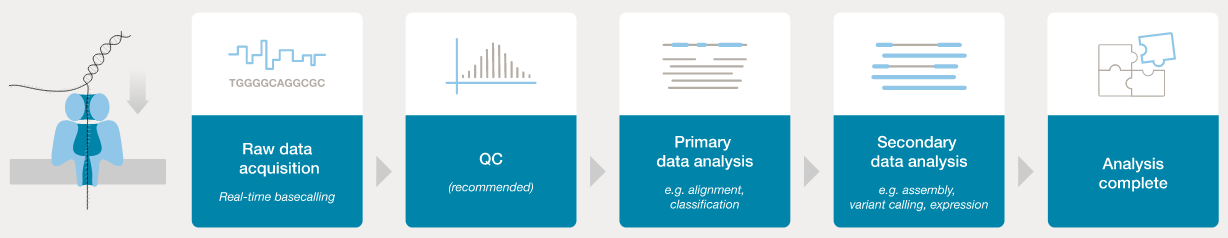
- ONT provides software (MinKNOW) for operating the MinION, and for generating the sequence data (e.g., the
guppybasecaller). - Once the raw FAST5 data have been converted to basecalls, we can use more familiar tools for quality assessment and analysis (e.g., FastQC).
https://nanoporetech.com/nanopore-sequencing-data-analysis
The current version of the Guppy software can be downloaded from the “Software Downloads” section of the Nanopore Community website:
https://community.nanoporetech.com/downloads
Note that you will need to create a login to access the website.
Today we will be using guppy_basecaller on the NeSI system to generate FASTQ data from a set of FAST5 files.
Recap: FASTQ data format
Assessing sequence quality: phred scores

Ewing B, Green P. (1998): Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8(3):186-194.

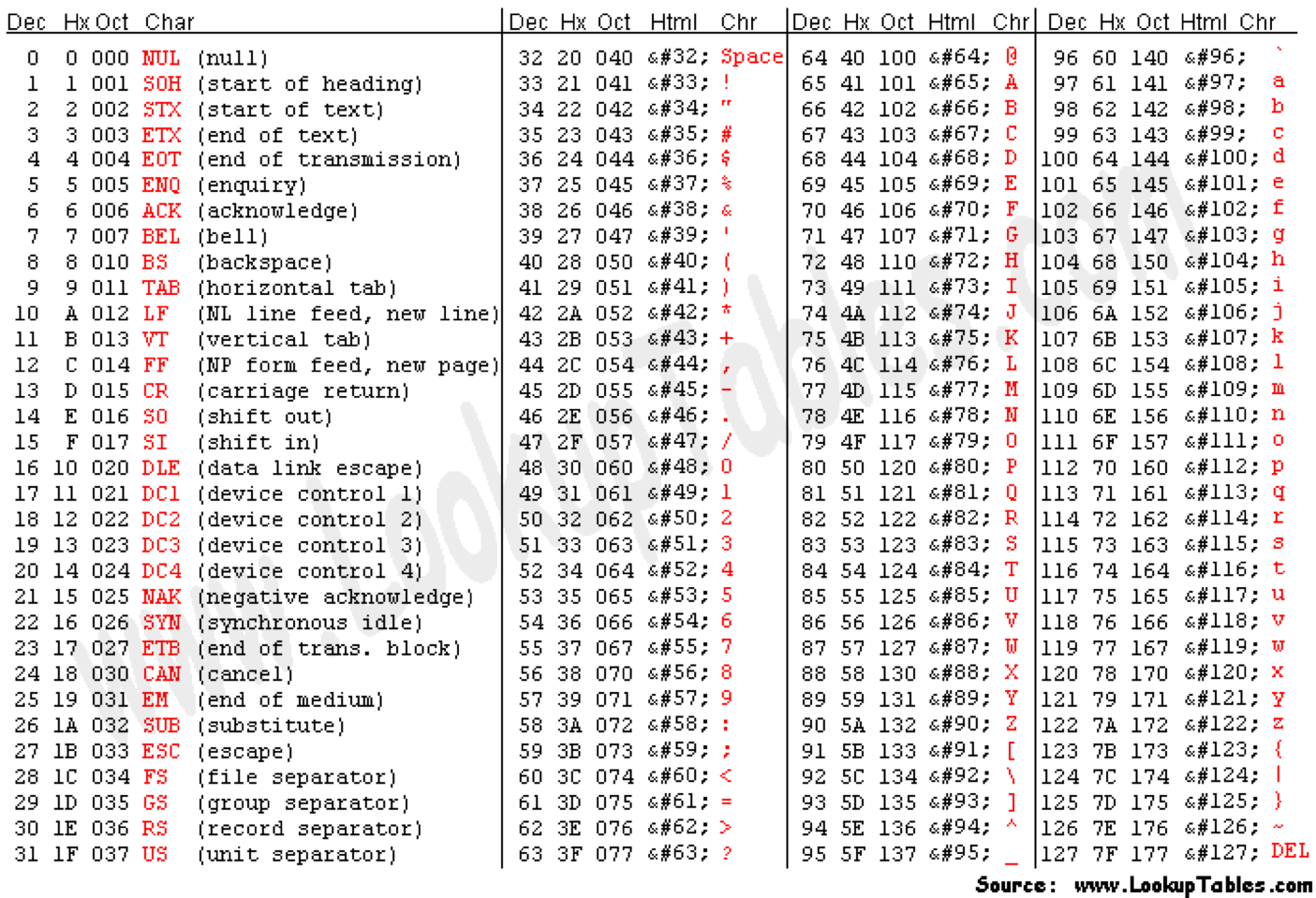
Can use ASCII to represent quality scores by adding 33 to the phred score and converting to ASCII.
- Quality score of 38 becomes 38+33=71: “G”
http://en.wikipedia.org/wiki/Phred_quality_score
- The FASTQ format allows the storage of both sequence and quality information for each read.
- This is a compact text-based format that has become the de facto standard for storing data from next generation sequencing experiments.
FASTQ format:

http://en.wikipedia.org/wiki/FASTQ_format
- Line 1: ‘@’ character followed by sequence identifier and optional description.
- Line 2: base calls.
- Line 3: ‘+’ character, optionally followed by the same sequence identifier (and description) again.
- Line 4: quality scores
E coli data set on NeSI
For this part of the workshop we are working at:
~/obss_2022/nanopore/ecoli-data/
Let’s change to that directory now:
cd ~/obss_2022/nanopore/ecoli-data/
FAST5 files from sequencing E. coli are available in the directory:
fast5_pass
ls -1 fast5_pass
ecoli_pass_0.fast5
ecoli_pass_10.fast5
ecoli_pass_11.fast5
ecoli_pass_12.fast5
ecoli_pass_13.fast5
ecoli_pass_14.fast5
ecoli_pass_15.fast5
ecoli_pass_1.fast5
ecoli_pass_2.fast5
ecoli_pass_3.fast5
ecoli_pass_4.fast5
ecoli_pass_5.fast5
ecoli_pass_6.fast5
ecoli_pass_7.fast5
ecoli_pass_8.fast5
ecoli_pass_9.fast5
Basecalling: guppy
guppyis a neural network based basecaller.- analyses the electrical trace data and predicts base
- it is GPU-aware, and can basecall in real time
- can also call base modifications (e.g., 5mC, 6mA)
- high accuracy mode (slower) and super-high accuracy mode (even slower) can improve basecalls post-sequencing
- The software is provided by ONT, and the source code is available via a developer agreement.
- Output is the standard “FASTQ” format for sequence data.
On NeSI:
module load ont-guppy-gpu/6.2.1
guppy_basecaller --help
: Guppy Basecalling Software, (C) Oxford Nanopore Technologies plc. Version 6.2.1+6588110, minimap2 version 2.22-r1101
Use of this software is permitted solely under the terms of the end user license agreement (EULA).By running, copying or accessing this software, you are demonstrating your acceptance of the EULA.
The EULA may be found in /scale_wlg_persistent/filesets/opt_nesi/CS400_centos7_bdw/ont-guppy-gpu/6.2.1/bin
Usage:
With config file:
guppy_basecaller -i <input path> -s <save path> -c <config file> [options]
With flowcell and kit name:
guppy_basecaller -i <input path> -s <save path> --flowcell <flowcell name>
--kit <kit name>
List supported flowcells and kits:
guppy_basecaller --print_workflows
Use GPU for basecalling:
guppy_basecaller -i <input path> -s <save path> -c <config file>
--device <cuda device name> [options]
Command line parameters:
[ TRUNCATED - this goes on for a long time... ]
Using guppy on NeSI
The guppy_basecaller software can use GPUs to speed up the basecalling process.
On NeSI, we can access the A100 GPUs by submitting a job to the cluster using a slurm script.
The slurm script we will use can be found at:
scripts/guppy_fastmodel.sl
Let’s have a look at the content:
more scripts/guppy_fastmodel.sl
#!/bin/bash -e
#SBATCH --job-name guppy_basecalling
#SBATCH --time 00:10:00
#SBATCH --mem 4G
#SBATCH --cpus-per-task 2
#SBATCH --output guppy_%j.out
#SBATCH --account nesi02659
#SBATCH --gpus-per-node A100-1g.5gb:1
module purge
module load ont-guppy-gpu/6.2.1
guppy_basecaller -i fast5_pass -s fastq_fastmodel \
--config /opt/nesi/CS400_centos7_bdw/ont-guppy-gpu/6.2.1/data/dna_r9.4.1_450bps_fast.cfg \
--device auto --recursive --records_per_fastq 4000 --min_qscore 7 --compress_fastq
- The
SBATCHcommands are providing information to theslurmjob scheduler: job name, maximum run time, memory allocation etc - The
modulecommands are making sure the necessary modules are available to the script (here we are specifying version 6.2.1 ofont-guppy-gpu- the GPU-enabled version of ONT’s guppy software). - The
guppy_basecallercommand is what is doing the work (i.e., converted the .fast5 data to .fastq).
Let’s have a closer look at the guppy_basecaller command:
guppy_basecaller \
-i fast5_pass_subset \
-s fastq_fastmodel \
--config /opt/nesi/CS400_centos7_bdw/ont-guppy-gpu/6.2.1/data/dna_r9.4.1_450bps_fast.cfg \
--device auto \
--recursive \
--records_per_fastq 4000 \
--min_qscore 7 \
--compress_fastq
Options used:
-i fast5_pass_subset: where to find the fast5 files (note the we are assuming that the script in run from the directory:~/obss_2022/nanopore/)-s fastq_fastmodel: the name of the folder where the .fastq files will be written to.-config /opt/nesi/CS400_centos7_bdw/ont-guppy-gpu/6.2.1/data/dna_r9.4.1_450bps_fast.cfg: this specifies the model that is being used to perform the basecalling. In this case we are using the “fast” model (rather than high-accuracy (hac) or super-high accuracy (sup)) to save time, anddnadenotes that we sequenced… DNA.r9.4.1refers to the flowcell version that was used to generate the data, and 450bps is the speed at which the DNA passes through the pore (450 bases per second).--device auto: this refers to which GPU device should be used (here we let the software selectautomatically, but it can sometimes be useful to select specific GPU devices).--recursive: search recursively for.fast5files (actually all our.fast5files are in the folder, so this isn’t needed here).--records_per_fastq 4000: number of reads to include per.fastqfile.--min_qscore 7: minimum average base quality score to considered a read to have “passed” QC.--compress_fastq: create compressed (.fastq.gz) files.
To submit the script, we use the sbatch command, and run it from the ~/obss_2022/nanopore/ecoli-data directory. You can check
if you are in that directory with pwd. If not: cd ~/obss_2022/nanopore/ecoli-data.
To run the script:
sbatch scripts/guppy_fastmodel.sl
Once the script has been submitted, you can check to make sure it is running via:
# This shows all your current jobs
squeue --me
JOBID USER ACCOUNT NAME CPUS MIN_MEM PARTITI START_TIME TIME_LEFT STATE NODELIST(REASON)
31430356 michael. nesi02659 spawner-jupy 2 4G infill 2022-11-20T0 1:23:52 RUNNING wbl004
31431766 michael. nesi02659 guppy_baseca 2 4G gpu 2022-11-20T0 6:28 RUNNING wbl001
It will also write progress to a log file that contains the job ID (represented by XXXXXXX in the code below).
You can check the file name using ls -ltr (list file details, in reverse time order).
Using the tail -f command, we can watch the progress (use control-c to exit):
ls -ltr
tail -f guppy_XXXXXXX.out
Example of output:
Use of this software is permitted solely under the terms of the end user license agreement (EULA).By running, copying or accessing this software, you are demonstrating your acceptance of the EULA.
The EULA may be found in /scale_wlg_persistent/filesets/opt_nesi/CS400_centos7_bdw/ont-guppy-gpu/6.2.1/bin
Found 16 fast5 files to process.
Init time: 847 ms
0% 10 20 30 40 50 60 70 80 90 100%
|----|----|----|----|----|----|----|----|----|----|
****************************
Use “Control-c” to exit (it’s okay, it won’t kill the job).
The job should take around 6 minutes to run.
Once the job has completed successfully, the .fastq files can be found at fastq_fastmodel:
ls -1 fastq_fastmodel
Note that pass and fail subfolders have been created.
ls -l fastq_fastmodel/pass/
ls -l fastq_fastmodel/fail/
Note that both directories (pass and fail) contain the same number of files, and they have exactly the same names. BUT, their content is different: the .fastq.gz files in the pass folder contain data for the reads that have passed QC (average base quality above threshold), and the fail folder contains those that failed. The files in the pass folder should be (hopefully!) much larger those their counterparts in the fail folder.
ls -lh fastq_fastmodel/pass/fastq_runid_15ca037c8ec4904bf5408d8ae5a1abecb8458aee_0_0.fastq.gz
-rw-rw----+ 1 michael.black nesi02659 5.7M Nov 20 00:58 fastq_fastmodel/pass/fastq_runid_15ca037c8ec4904bf5408d8ae5a1abecb8458aee_0_0.fastq.gz
ls -lh fastq_fastmodel/fail/fastq_runid_15ca037c8ec4904bf5408d8ae5a1abecb8458aee_0_0.fastq.gz
-rw-rw----+ 1 michael.black nesi02659 672K Nov 20 00:58 fastq_fastmodel/fail/fastq_runid_15ca037c8ec4904bf5408d8ae5a1abecb8458aee_0_0.fastq.gz
Here the first .fastq.gz file (in the pass folder) contains 5.7M of data, whereas the one in the fail folder only contains 672K (i.e., it is about 1/12 of the size).
Key Points
FAST5 data can be converted to FASTQ via the guppy_basecaller software.
guppy_basecaller has different models (FAST, HAC and SUP) that provide increasing basecall accuracy (and take increasing amounts of computation).
Once bascalling has been done, standard tasks such as QA/QC, mapping and variant calling can be performed.